%%html
<script src="https://bits.csb.pitt.edu/preamble.js"></script>
Sequence Project¶
https://MSCBIO.github.io/files/hydra179.aln
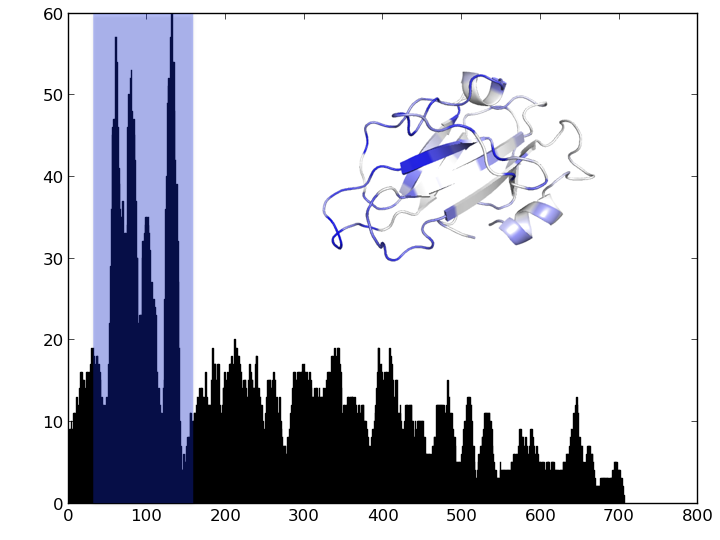
Identify the region of this gene with the most variation.
- For each position, identify and plot the number of unique residues
- For each position, identify and plot the number of unique subsequences of length 10
import matplotlib.pyplot as plt
from Bio import AlignIO
def countatpos(seqs,pos):
'''count the number of unique subsequences of length 10 at pos in seqs'''
vals = set()
for s in seqs:
vals.add(str(s[pos:pos+10].seq))
return len(vals)
seqs = AlignIO.read("../files/hydra179.aln",'clustal');
xaxis = list()
yaxis = list()
for i in range(seqs.get_alignment_length()):
xaxis.append(i)
yaxis.append(countatpos(seqs,i))
plt.bar(xaxis,yaxis,width=1);

A bit more python... list comprehensions¶
A concise way to create lists
[ <expr of var> for <var> in <iterable> if <condition> ]
[x for x in range(10) if x % 2 == 0]
[0, 2, 4, 6, 8]
Three ways to do the same thing¶
squares = [x**2 for x in range(10)]
squares
[0, 1, 4, 9, 16, 25, 36, 49, 64, 81]
squares = []
for x in range(10):
squares.append(x**2)
squares
[0, 1, 4, 9, 16, 25, 36, 49, 64, 81]
squares = list(map(lambda x: x**2, range(10)))
squares
[0, 1, 4, 9, 16, 25, 36, 49, 64, 81]
Other comprehensions¶
list(enumerate('ABCD')) #enumerate returns tuples of index,value
[(0, 'A'), (1, 'B'), (2, 'C'), (3, 'D')]
Dictionary comprehension¶
{key: val for key, val in enumerate('ABCD') if val not in 'CB'}
{0: 'A', 3: 'D'}
Set comprehension¶
{v for v in 'ABCDABCD' if v not in 'CB'}
{'A', 'D'}
Should you use comprehensions?¶
Sure, if the result is short and easy to understand
But don't go out of your way to use them - they can get ugly fast
result = [line.strip().split("\t") for line in open("file") if not line.startswith('#')]
%%html
<div id="seq2comp" style="width: 500px"></div>
<script>
$('head').append('<link rel="stylesheet" href="https://bits.csb.pitt.edu/asker.js/themes/asker.default.css" />');
var divid = '#seq2comp';
jQuery(divid).asker({
id: divid,
question: "What is in result?",
answers: ["A","B","C","D","E"],
extra: ['A list of lists of the tab separated values of lines with #',
'A list of lists of the tab separated values of lines without #',
'A list of the lines without #',
'A list of the tab separated values of lines without #',
'I have no idea'],
server: "https://bits.csb.pitt.edu/asker.js/example/asker.cgi",
charter: chartmaker})
$(".jp-InputArea .o:contains(html)").closest('.jp-InputArea').hide();
</script>
seqs = AlignIO.read("../files/hydra179.aln",'clustal');
yaxis = [len({str(s[i:i+10].seq) for s in seqs}) for i in range(seqs.get_alignment_length())]
plt.bar(range(seqs.get_alignment_length()),yaxis,width=1);
Back to Biopython...¶
from Bio import AlignIO
a = AlignIO.read('../files/hydra179.aln','clustal')
len(a)
179
len(a[0]),a.get_alignment_length()
(706, 706)
a
<<class 'Bio.Align.MultipleSeqAlignment'> instance (179 records of length 706) at 1199dbd00>
Phylogenetic Trees¶
A phylogenetic tree or evolutionary tree is a branching diagram or "tree" showing the inferred evolutionary relationships among various biological species or other entities—their phylogeny—based upon similarities and differences in their physical or genetic characteristics. --Wikipedia
Biopython can read a variety of tree formats: Newick (clustal), NEXUS, phyloXML, NeXML, and CDAO.
from Bio import Phylo
tree = Phylo.read('../files/hydra179.dnd','newick') #must specify format
tree
Tree(rooted=False, weight=1.0)
Displaying trees¶
Phylo.draw_ascii(tree)
______ gi|302171738|gb|ADK97770.1|
____|
| |____ gi|302171740|gb|ADK97771.1|
_____|
| | , gi|313105485|gb|ADR32101.1|
| |_______________________________|
| | gi|313105490|gb|ADR32105.1|
|
| , gi|225423246|gb|ACN91129.1|
| |
| | gi|407380197|gb|AFU11414.1|
| |
| |, gi|302171754|gb|ADK97778.1|
| ||
| || gi|407380047|gb|AFU11341.1|
| |
| ,, gi|407380097|gb|AFU11366.1|
| ||
| || gi|407380101|gb|AFU11368.1|
| ||
| || gi|407380017|gb|AFU11326.1|
| ||
| || gi|407380135|gb|AFU11385.1|
| ||
| || gi|407380117|gb|AFU11376.1|
| |
| |, gi|407380023|gb|AFU11329.1|
| ||
| || gi|407380045|gb|AFU11340.1|
| ||
| || gi|302171762|gb|ADK97782.1|
| |
| |_ gi|407380093|gb|AFU11364.1|
| |
| | , gi|302171792|gb|ADK97797.1|
| | |
| |,| gi|302171794|gb|ADK97798.1|
| |||
| ||| gi|225423258|gb|ACN91135.1|
| ||
| ,| gi|407380183|gb|AFU11407.1|
| ||
| || gi|302171760|gb|ADK97781.1|
| |
| |__ gi|225423252|gb|ACN91132.1|
| |
| | , gi|407380005|gb|AFU11320.1|
| | |
| |_| gi|407380157|gb|AFU11396.1|
| | |
| | | gi|407380057|gb|AFU11346.1|
| |
| | , gi|302171782|gb|ADK97792.1|
| | |
| | | gi|407380151|gb|AFU11393.1|
| | |
__| |,| gi|407380113|gb|AFU11374.1|
| | ,|||
| | |||| gi|407380085|gb|AFU11360.1|
| | |||
| | |||, gi|407380139|gb|AFU11387.1|
| | ||||
| | |||| gi|407380181|gb|AFU11406.1|
| | |||
| | ||| gi|407380035|gb|AFU11335.1|
| | |||
| | |||_ gi|407380123|gb|AFU11379.1|
| | |||
| | ||| gi|407380187|gb|AFU11409.1|
| | ||
| | ||, gi|225423250|gb|ACN91131.1|
| | |||
| | |,| gi|407380176|gb|AFU11404.1|
| | |||
| | ||| gi|407380103|gb|AFU11369.1|
| | ||
| | || gi|407379993|gb|AFU11314.1|
| | |
| | | , gi|225423248|gb|ACN91130.1|
| | | |
| | | | gi|225423296|gb|ACN91154.1|
| | | |
| | | | gi|225423298|gb|ACN91155.1|
| | | |
| | | ,| gi|302171786|gb|ADK97794.1|
| | | ||
| | |,|| gi|302171788|gb|ADK97795.1|
| | |||
| | ||| gi|407380051|gb|AFU11343.1|
| | ||
| | || , gi|225423280|gb|ACN91146.1|
| | || |
| | ||,| gi|302171758|gb|ADK97780.1|
| | ||||
| | |||| gi|407380185|gb|AFU11408.1|
| | | |
| | | |, gi|407380007|gb|AFU11321.1|
| | | ||
| | | | gi|407380105|gb|AFU11370.1|
| | |
| | |_ gi|407380133|gb|AFU11384.1|
| | |
,| | | , gi|225423276|gb|ACN91144.1|
|| | | |
|| | | | gi|407380001|gb|AFU11318.1|
|| | | |
|| | | | gi|302171752|gb|ADK97777.1|
|| | | |
|| | | | gi|313105489|gb|ADR32104.1|
|| | | |
|| |___| | gi|407380003|gb|AFU11319.1|
|| | |
|| | , gi|225423300|gb|ACN91156.1|
|| | |
|| | | gi|302171790|gb|ADK97796.1|
|| | |
|| | ,| gi|407380031|gb|AFU11333.1|
|| | ||
|| | ||, gi|407380077|gb|AFU11356.1|
|| | |,|
|| | ||| gi|407380079|gb|AFU11357.1|
|| | ||
|| | || gi|407380015|gb|AFU11325.1|
|| | |
|| | |, gi|302171764|gb|ADK97783.1|
|| |,||
|| |||| gi|407380171|gb|AFU11402.1|
|| ||,|
|| |||, gi|407380033|gb|AFU11334.1|
|| ||||
|| |||| gi|407380083|gb|AFU11359.1|
|| |||
|| |||_ gi|407380013|gb|AFU11324.1|
|| |||
|| ||| , gi|407380037|gb|AFU11336.1|
|| |||,|
,|| |||| gi|407380039|gb|AFU11337.1|
||| |,|
||| ||, gi|407380173|gb|AFU11403.1|
||| |||
||| ||| gi|407380210|gb|AFU11420.1|
||| ||
||| ||_ gi|407380075|gb|AFU11355.1|
||| ||
||| || gi|407380189|gb|AFU11410.1|
||| |
||| |_ gi|407380143|gb|AFU11389.1|
|||
|||__ gi|302171734|gb|ADK97768.1|
|||
|||___ gi|302171736|gb|ADK97769.1|
||
|| , gi|225423290|gb|ACN91151.1|
|| |
|| | gi|407380025|gb|AFU11330.1|
|| |
|| | gi|302171774|gb|ADK97788.1|
|| |
||,| gi|225423244|gb|ACN91128.1|
||||
||||, gi|407380155|gb|AFU11395.1|
|,|||
||| | gi|407380179|gb|AFU11405.1|
|||
||| gi|225423272|gb|ACN91142.1|
||
|| , gi|407380061|gb|AFU11348.1|
||,|
|||| gi|407380205|gb|AFU11418.1|
|||
|||, gi|407380069|gb|AFU11352.1|
||||
|| | gi|407380195|gb|AFU11413.1|
||
||_ gi|407380019|gb|AFU11327.1|
||
||_ gi|407380053|gb|AFU11344.1|
|
| , gi|225423284|gb|ACN91148.1|
| |
|_| gi|407380109|gb|AFU11372.1|
| |
| | gi|302171750|gb|ADK97776.1|
|
| , gi|302171784|gb|ADK97793.1|
| |
| | gi|407380089|gb|AFU11362.1|
| |
|,| gi|225423254|gb|ACN91133.1|
|||
,|| gi|225423294|gb|ACN91153.1|
||
|| gi|407379991|gb|AFU11313.1|
|
|, gi|225423260|gb|ACN91136.1|
||
|| gi|407380131|gb|AFU11383.1|
||
||, gi|407380027|gb|AFU11331.1|
|||
||| gi|407380029|gb|AFU11332.1|
|||
|,| gi|302171778|gb|ADK97790.1|
|||
||| gi|225423266|gb|ACN91139.1|
||
||, gi|225423292|gb|ACN91152.1|
|||
||| gi|302171776|gb|ADK97789.1|
||
|, gi|407380055|gb|AFU11345.1|
||
|| gi|407380214|gb|AFU11422.1|
||
||, gi|407380041|gb|AFU11338.1|
|||
||| gi|407380149|gb|AFU11392.1|
|||
||| gi|407380169|gb|AFU11401.1|
|||
,|| gi|225423268|gb|ACN91140.1|
||
||, gi|302171770|gb|ADK97786.1|
|,|
||| gi|407380203|gb|AFU11417.1|
||
||, gi|407379997|gb|AFU11316.1|
|||
||| gi|407380009|gb|AFU11322.1|
|||
||| gi|407380141|gb|AFU11388.1|
|||
||| gi|407380011|gb|AFU11323.1|
||
|, gi|407380021|gb|AFU11328.1|
||
|| gi|407380191|gb|AFU11411.1|
||
|| gi|407379989|gb|AFU11312.1|
|
|_ gi|407380095|gb|AFU11365.1|
|
| , gi|225423270|gb|ACN91141.1|
| ,|
|,|| gi|407380193|gb|AFU11412.1|
|||
||| gi|407380147|gb|AFU11391.1|
||
||, gi|225423288|gb|ACN91150.1|
|||
||| gi|302171768|gb|ADK97785.1|
|||
||, gi|407380043|gb|AFU11339.1|
|,|
||| gi|407380111|gb|AFU11373.1|
|||
||| gi|407380212|gb|AFU11421.1|
||
|| gi|407380067|gb|AFU11351.1|
||
|| gi|302171756|gb|ADK97779.1|
||
||, gi|407380063|gb|AFU11349.1|
|,|
||| gi|407380145|gb|AFU11390.1|
||
|| gi|407380160|gb|AFU11397.1|
|
|, gi|302171746|gb|ADK97774.1|
||
|| gi|407380137|gb|AFU11386.1|
|
|, gi|225423286|gb|ACN91149.1|
||
|| gi|302171766|gb|ADK97784.1|
||
|| gi|225423256|gb|ACN91134.1|
||
|| gi|407380164|gb|AFU11399.1|
||
|| gi|407380199|gb|AFU11415.1|
||
|, gi|225423282|gb|ACN91147.1|
||
|| gi|302171748|gb|ADK97775.1|
||
,| gi|407379995|gb|AFU11315.1|
||
|| gi|407379987|gb|AFU11311.1|
||
|| gi|407380065|gb|AFU11350.1|
|
|, gi|302171772|gb|ADK97787.1|
||
|| gi|407380127|gb|AFU11381.1|
,|
|, gi|407380099|gb|AFU11367.1|
||
|| gi|407380129|gb|AFU11382.1|
|
|, gi|407380119|gb|AFU11377.1|
||
|| gi|407380121|gb|AFU11378.1|
||
|| gi|407380081|gb|AFU11358.1|
|
, gi|302171780|gb|ADK97791.1|
|
| gi|407380073|gb|AFU11354.1|
|
, gi|407380107|gb|AFU11371.1|
|
| gi|407380153|gb|AFU11394.1|
|
, gi|407380115|gb|AFU11375.1|
|
| gi|407380201|gb|AFU11416.1|
|
| ___ model1_F.pdb
_| _|
|,| | gi|407380166|gb|AFU11400.1|
|||
||| gi|407380162|gb|AFU11398.1|
||
|| gi|407379985|gb|AFU11310.1|
||
,| gi|407380059|gb|AFU11347.1|
||
|| gi|164609123|gb|ABY62783.1|
|
| gi|225423262|gb|ACN91137.1|
|
| gi|407380049|gb|AFU11342.1|
|
| gi|407380208|gb|AFU11419.1|
|
, gi|407380071|gb|AFU11353.1|
|
| gi|407380091|gb|AFU11363.1|
|
|, gi|225423274|gb|ACN91143.1|
||
|| gi|225423278|gb|ACN91145.1|
||
|| gi|302171742|gb|ADK97772.1|
||
,| gi|302171744|gb|ADK97773.1|
||
|| gi|313105486|gb|ADR32102.1|
|
| gi|225423264|gb|ACN91138.1|
|
, gi|407379999|gb|AFU11317.1|
|
| gi|407380087|gb|AFU11361.1|
|
| gi|407380125|gb|AFU11380.1|
Displaying trees¶
Phylo can draw trees using matplot lib (e.g., can use savefig etc)
%matplotlib inline
Phylo.draw(tree)

Phylo.draw(tree,label_func=lambda x: None)

Motifs¶
Sequence motifs are short, recurring patterns in DNA that are presumed to have a biological function. Often they indicate sequence-specific binding sites for proteins such as nucleases and transcription factors (TF). --The Internet
Motif logos¶
from Bio import motifs #lower case for some reason
m = motifs.create(["TACAA","CATGC","TACTA","CCCAA"])
m.counts
{'A': [0, 3, 0, 2, 3],
'C': [2, 1, 3, 0, 1],
'G': [0, 0, 0, 1, 0],
'T': [2, 0, 1, 1, 0]}
m.consensus
Seq('CACAA')
m.weblogo('logo.png',alphabet='alphabet_dna',stack_width='large')
from IPython.display import Image
Image(filename='./logo.png')

Reading Motifs¶
f = open('../files/MA0004.1.sites') # unlike other parts of Biopython, can't just provide filename to open
arnt = motifs.read(f,'sites') #JASPAR sites
arnt
<Bio.motifs.jaspar.Motif at 0x119703d00>
arnt.consensus
Seq('CACGTG')
print(arnt.counts)
0 1 2 3 4 5 A: 4.00 19.00 0.00 0.00 0.00 0.00 C: 16.00 0.00 20.00 0.00 0.00 0.00 G: 0.00 1.00 0.00 20.00 0.00 20.00 T: 0.00 0.00 0.00 0.00 20.00 0.00
arnt.instances
[Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('AACGTG'),
Seq('AACGTG'),
Seq('AACGTG'),
Seq('AACGTG'),
Seq('CGCGTG')]
Scoring Matrices¶
The counts attribute can be normalized to represent probabilities at each position
print(arnt.counts.normalize())
0 1 2 3 4 5 A: 0.20 0.95 0.00 0.00 0.00 0.00 C: 0.80 0.00 1.00 0.00 0.00 0.00 G: 0.00 0.05 0.00 1.00 0.00 1.00 T: 0.00 0.00 0.00 0.00 1.00 0.00
A pseudocount is often added at each position to prevent probability from going to zero.
print(arnt.counts.normalize(pseudocounts=0.8))
0 1 2 3 4 5 A: 0.21 0.85 0.03 0.03 0.03 0.03 C: 0.72 0.03 0.90 0.03 0.03 0.03 G: 0.03 0.08 0.03 0.90 0.03 0.90 T: 0.03 0.03 0.03 0.03 0.90 0.03
PSSM¶
The position-specific scoring matrix is the position weight matrix (with pseudocounts) expressed as a log (base 2) odds ratios
pwm = arnt.counts.normalize(pseudocounts=0.8)
pssm = pwm.log_odds()
print(pssm)
0 1 2 3 4 5 A: -0.27 1.77 -2.86 -2.86 -2.86 -2.86 C: 1.53 -2.86 1.84 -2.86 -2.86 -2.86 G: -2.86 -1.69 -2.86 1.84 -2.86 1.84 T: -2.86 -2.86 -2.86 -2.86 1.84 -2.86
A negative value means a nucleotide is less likely than the background at a specific position.
By default a uniform background is assumed, but this can be changed with the background parameter of log_odds.
%%html
<div id="logpssm" style="width: 500px"></div>
<script>
$('head').append('<link rel="stylesheet" href="https://bits.csb.pitt.edu/asker.js/themes/asker.default.css" />');
var divid = '#logpssm';
jQuery(divid).asker({
id: divid,
question: "What is the PSSM score for A at a position if it occurs 5 times out of 20 sequences (no pseudocount)?",
answers: ["-1","-.25","0",".25","1","None of the above"],
server: "https://bits.csb.pitt.edu/asker.js/example/asker.cgi",
charter: chartmaker})
$(".jp-InputArea .o:contains(html)").closest('.jp-InputArea').hide();
</script>
Search for Motifs¶
from Bio import SeqIO
from Bio.Seq import Seq
largeseq = SeqIO.read('../files/bnip3.fasta','fasta') #load with same alphabet as motif
smallseq = Seq('AAACCCACGTGACTATATA')
We can search for exact matches
for pos,seq in arnt.instances.search(smallseq):
print("%i %s" % (pos,seq))
5 CACGTG
for pos,seq in arnt.instances.search(largeseq.seq): # pass sequence, not seqrecord
print("%i %s" % (pos,seq))
3452 CACGTG 4058 CACGTG 6181 AACGTG 8591 CGCGTG 10719 CACGTG 10998 CACGTG
Searching for Motifs¶
results = [(pos,str(seq)) for pos,seq in arnt.instances.search(largeseq.seq)]
len(results)
6
print(results)
[(3452, 'CACGTG'), (4058, 'CACGTG'), (6181, 'AACGTG'), (8591, 'CGCGTG'), (10719, 'CACGTG'), (10998, 'CACGTG')]
arnt.instances
[Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('CACGTG'),
Seq('AACGTG'),
Seq('AACGTG'),
Seq('AACGTG'),
Seq('AACGTG'),
Seq('CGCGTG')]
Searching for Motifs¶
We can use the PSSM to get a fuzzier match
pwm = arnt.counts.normalize(pseudocounts=0.8)
pssm = pwm.log_odds()
positions = [pos for pos,seq in pssm.search(largeseq.seq)]
len(positions)
1065
results = [(pos,score) for pos,score in pssm.search(largeseq.seq, threshold=4)]
len(results)
167
The score is a $log_2$ likelihood so a score of 4 is $2^4=16$ times more likely to occur as part of the motif than as part of the (uniform) background
results[0]
(508, 5.975107)
Searching for Motifs¶
Positions may be negative if the motif was found on the reverse strand.
results[:2]
[(508, 5.975107), (-13823, 6.0461903)]
pos = results[1][0] #-13823
hit = largeseq.seq[pos:pos+len(arnt)] #negative indices can still be used to retrieve matched subsequence
print(pos,len(largeseq)+pos)
print(hit, hit.reverse_complement())
-13823 508 CACGGG CCCGTG
print(arnt.counts)
0 1 2 3 4 5 A: 4.00 19.00 0.00 0.00 0.00 0.00 C: 16.00 0.00 20.00 0.00 0.00 0.00 G: 0.00 1.00 0.00 20.00 0.00 20.00 T: 0.00 0.00 0.00 0.00 20.00 0.00
Some more marine biology¶

Your Herculean Task¶
0. Get input files of 179 hydra sequences
!wget https://MSCBIO2025.github.io/files/hydra179.aln
!wget https://MSCBIO2025.github.io/files/hydra179.dnd
!wget https://MSCBIO2025.github.io/files/hydra179.fasta
- Display the phylogenetic tree from the clustal alignment (hydra179.dnd)
- Identify the subsequence of length 20 that has the most variation amount these sequences (like last time)
- Use clustal to compute the multiple alignment of these 179 length 20 subsequences
- Display the phylogenetic tree from this alignment
#!/usr/local/bin/python
from Bio import SeqIO
from Bio import AlignIO
from Bio.Seq import Seq
from Bio import Phylo
from Bio.Align.Applications import ClustalwCommandline
import sys
sys.argv = ['fake','../files/hydra179.aln']
a = AlignIO.read(sys.argv[1],'clustal')
#the following is perhaps not the most readable, but it counts
#the number of unique sequences of length 20 at each position in align
cnts = [len({str(s.seq) for s in a[:,i:i+20]}) for i in range(len(a[0])-20)]
maxpos = cnts.index(max(cnts))
maxseqs = a[:,maxpos:maxpos+20]
# have to write out to a file to compute msa with clustal
out = open('subalign.fasta','w')
SeqIO.write(maxseqs, out, 'fasta')
out.close() # must close before reading
cline = ClustalwCommandline("clustalw", infile="subalign.fasta", outfile='subalign.aln')
# cline()
# msa of the subsequences
maxalign = AlignIO.read('subalign.aln','clustal')
tree = Phylo.read('subalign.dnd','newick')
tree.root.branch_length = 0 #prettier
Phylo.draw(tree,label_func=lambda x: None)
