%%html
<script src="https://bits.csb.pitt.edu/preamble.js"></script>
Cheminformatics (also known as chemoinformatics, chemioinformatics and chemical informatics) is the use of computer and informational techniques applied to a range of problems in the field of chemistry. --Wikipedia
Open Source Cheminformatics¶
rdkit http://www.rdkit.org
- BSD License
- Relatively new, very nicely architected C++ backend
- Actively developed
- Native Python interface
OpenBabel http://openbabel.org
- GNU License
- Older (forked from OpenEye in 2001), a bit crufty and complicated
- Lots of functionality (e.g., support for more than 100 file formats)
- Python interface is through SWIG (auto-generated) bindings to C/C++
- Includes standalone programs: babel, obabel, etc.
Pybel
- A native, user-friendly python interface to OpenBabel
- Limited functionality (but can always fallback to OpenBabel)
- Simplest to use
- Note: Pybel is installed as part of openbabel. There is a completely unrelated python package called PyBEL that is not what you want
File Formats¶
2D
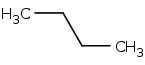 SMILES
SMILES
3D
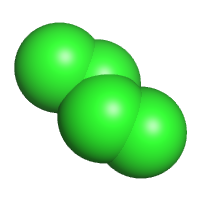 pdb, sdf, mol2
pdb, sdf, mol2
Simplified Molecular Input Line Entry System (SMILES)¶
Atoms
Specified by their atomic symbols inside brackets
- [Au], [Fe], [Zn], etc
No brackets needed for organic subset: B, C, N, O, P, S, F, Cl, Br, and I
Aromatic atoms are lower case: c1ccccc1
Bonds
- Single -
- Double =
- Triple #
- Aromatic :
Single and aromatic can be omitted.
%%html
<div id="chem1" style="width: 500px"></div>
<script>
var divid = '#chem1';
jQuery(divid).asker({
id: divid,
question: "What is a SMILES string for ethane?",
answers: ['cc','CC','C=C','C#C','[Ca]-[Ca]'],
server: "https://bits.csb.pitt.edu/asker.js/example/asker.cgi",
charter: chartmaker})
$(".jp-InputArea .o:contains(html)").closest('.jp-InputArea').hide();
</script>
SMILES, cont.¶
%%html
<div id="chem3" style="width: 500px"></div>
<script>
var divid = '#chem3';
jQuery(divid).asker({
id: divid,
question: "What is a SMILES expression for benzene?",
answers: ['c1cccc1','cccccc1','c2ccccc2','1cccccc1','c1ccccc1c'],
server: "https://bits.csb.pitt.edu/asker.js/example/asker.cgi",
charter: chartmaker})
$(".jp-InputArea .o:contains(html)").closest('.jp-InputArea').hide();
</script>
Drawing¶
All but the simplest smiles can be challenging to interpret (especially if chirality is included). Fortunately, you can use pybel (or molecular viewers like MarvinView) to convert them to their 2D representation.
Example: CC(NC1=CC=C(O)C=C1)=O
from openbabel import pybel
mol = pybel.readstring('smi','CC(NC1=CC=C(O)C=C1)=O')
mol

mol.draw(filename="imgs/accet.png",show=False)
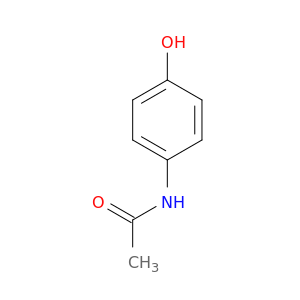
SMARTS¶
Regular expressions for molecules.
All SMILES are SMARTS (exact matches). Additionally, SMARTS support
- wild cards
C~*~Cany atom can be between two carbons using any (~) bonda1aaaaa1any aromatic 6 atom ring
- property testing
[R]atom in a ring[#6]atomic number is 6 (matches aromatic or aliphatic)[D3]atom with three explicit bonds (degree)
- logical operators (not - !, and - & ;, or - ,)
[!C&R]not aliphatic carbon and in ring[F,Cl,Br,I]one of the first four halogens
- matching an atomic environment ('recursive' SMARTS)
[$(*O);$(*C)]this matches one atom that is bound to both C and O
%%html
<div id="chem4" style="width: 500px"></div>
<script>
var divid = '#chem4';
jQuery(divid).asker({
id: divid,
question: "What is the best SMARTS expression for matching a 6 carbon ring?",
answers: ['[#6]1~[#6]~[#6]~[#6]~[#6]~[#6]1','c1ccccc1','[R][R][R][R][R][R]','[R]1[R][R][R][R][R]1','CCCCCC'],
server: "https://bits.csb.pitt.edu/asker.js/example/asker.cgi",
charter: chartmaker})
$(".jp-InputArea .o:contains(html)").closest('.jp-InputArea').hide();
</script>
Pybel Input/Output¶
pybel.readstring
Takes a format and string with molecular data in it and returns a single molecule.
mol = pybel.readstring('smi','CCCC')
len(mol.atoms)
4
For simple output, use the molecule's write method, which takes the format
mol.write('sdf','output.sdf',overwrite=True) #write to file
print(mol.write('sdf')) #no filename - return string
OpenBabel10302320482D
4 3 0 0 0 0 0 0 0 0999 V2000
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0 0 0 0
2 3 1 0 0 0 0
3 4 1 0 0 0 0
M END
$$$$
============================== *** Open Babel Warning in WriteMolecule No 2D or 3D coordinates exist. Stereochemical information will be stored using an Open Babel extension. To generate 2D or 3D coordinates instead use --gen2D or --gen3D.
pybel.readfile¶
pybel.readfile
Takes a format and file name and returns an iterator over all the molecules in the file.
mols = list(pybel.readfile('smi','../files/results.smi')) #expand the iterator into a list
len(mols)
14
mol = next(pybel.readfile('smi','../files/results.smi')) #get just first molecule
mol

#of course, this is the most efficient way to read all
for mol in pybel.readfile('smi','../files/results.smi'):
print(mol.write('can').rstrip()) #canonical smiles
N#Cc1c(O)c2C(=O)c3ccccc3C(=O)c2c(c1C#N)O NSC27034 N#Cc1cc2SCCSCCCSCCSc2cc1C#N NSC680721 N#Cc1cc2CN(CCN(CCN(CCN(Cc2cc1C#N)S(=O)(=O)c1ccc(cc1)C)S(=O)(=O)c1ccc(cc1)C)S(=O)(=O)c1ccc(cc1)C)S(=O)(=O)c1ccc(cc1)C NSC673657 N#Cc1cc2/C(=N\c3cccc(n3)N)/N=C(c2cc1C#N)Nc1cccc(n1)N NSC666078 N#Cc1c(OC)ccc(c1C#N)O.COc1ccc(c(c1C#N)C#N)OC NSC618324 N#Cc1c(C#N)c(O)c2c(c1O)c(N)ccc2 NSC320651 N#Cc1cc(ccc1C#N)NC(=O)CCCC(=O)Nc1ccc(c(c1)C#N)C#N NSC309816 N#Cc1c(C#N)c(O)c(c(c1O)Cl)Cl NSC172566 N#Cc1c(C#N)c(O)c2c(c1O)cccc2 NSC128281 N#Cc1cc(ccc1C#N)[N+](=O)[O-] NSC123374 N#Cc1cc(ccc1C#N)Oc1ccc(c(c1)C#N)C#N NSC94808 N#Cc1c2c(cc(c1C#N)[N+](=O)[O-])n(c1c2cccc1)C NSC92934 N#Cc1c(O)ccc(c1C#N)O NSC43554 N#Cc1ccccc1C#N NSC17562
pybel.Outputfile¶
To output many molecules to the same file, use pybel.Outputfile
output = pybel.Outputfile('sdf','output.sdf',overwrite=True)
for m in mols:
output.write(m)
Molecules¶
The molecule object provides a number of methods and access to the molecules atoms and bonds.
for atom in mol:
print(atom.atomicnum,end=' ')
6 6 6 6 6 6 6 7 6 7
Atom properties in clude atomicmass, atomicnum, coords, formalcharge, hyb, isotope, partialcharge, degree, explicitvalence and totalvalence
Atoms can also be accessed in mol.atoms
%%html
<div id="chemval" style="width: 500px"></div>
<script>
var divid = '#chemval';
jQuery(divid).asker({
id: divid,
question: "What is the valence of the first atom of CC?",
answers: ['0','1','2','3','4'],
server: "https://bits.csb.pitt.edu/asker.js/example/asker.cgi",
charter: chartmaker})
$(".jp-InputArea .o:contains(html)").closest('.jp-InputArea').hide();
</script>
SMARTS Matching¶
SMARTS matching is done by initializing a pybel.Smarts object with a SMARTS expression. This can then be applied to any molecule to identify the matching atoms.
mol

aromatic_ring = pybel.Smarts('a1aaaaa1')
aromatic_ring.findall(mol) #returns all _unique_ matches
[(1, 6, 5, 4, 3, 2)]
The returned matches are atom indices that can be accessed through mol.atoms
double_ring = pybel.Smarts('a1aaaa2a1aaaa2')
for (i,m) in enumerate(mols):
if double_ring.findall(m):
print(i)
m.draw(filename="r%d.png"%i,show=False)
5 8
mols[5]

mols[8]

Molecular Properties¶
print(mol.molwt) #molecular weight
128.13076
desc = mol.calcdesc()
print(desc)
{'abonds': 6.0, 'atoms': 10.0, 'bonds': 10.0, 'cansmi': nan, 'cansmiNS': nan, 'dbonds': 0.0, 'formula': nan, 'HBA1': 2.0, 'HBA2': 2.0, 'HBD': 0.0, 'InChI': nan, 'InChIKey': nan, 'L5': nan, 'logP': 1.42996, 'MP': 79.79220000000001, 'MR': 35.872, 'MW': 128.13076, 'nF': 0.0, 'rotors': 0.0, 's': nan, 'sbonds': 2.0, 'smarts': nan, 'tbonds': 2.0, 'title': nan, 'TPSA': 47.58}
desc['logP'] #calculated partition coefficient between octanol/water
1.42996
Lipinski's Rule of Five¶
In 1997 Christopher Lipinski analyzed existing drugs and came up with a set of molecular property rules for classifying a small molecule as drug-like.
- No more than 5 hydrogen bond donors
- No more than 10 hydrogen bond acceptors
- Molecular weight less than 500 daltons
- Partition coefficient logP less than 5
- There is no fifth rule
def lipinski(mol):
desc = mol.calcdesc()
return desc['HBD'] <= 5 and desc['HBA1'] <= 10 and desc['MW'] <= 500 and desc['logP'] <= 5
for m in mols:
print(lipinski(m))
True True False True True True True True True True True True True True
Fingerprints¶
A molecular fingerprint reduces the chemical features of a molecule into a bit vector. The features of the fingerprint correspond to a bit in the vector. This bit is set if the compound has that feature.
The most common type of fingerprint is a Daylight style fingerprint where all the paths (up to a given length) are enumerated and hashed to their bit positions.
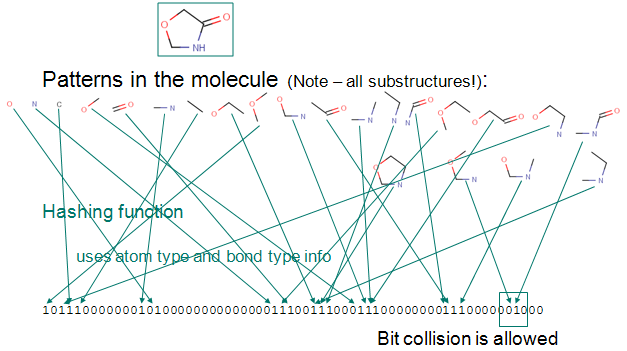
Fingerprints, cont.¶
Bit vectors can easily be compared, most commonly with the Tanimoto coefficient: $$\frac{A \cap B}{A \cup B}$$
This provides a quantitative measure of chemical similarity.
Similarity search is a surprisingly effective mechanism of virtual screening (given enough data).
fp = mol.calcfp()
type(fp)
openbabel.pybel.Fingerprint
print(fp.bits)
[75, 82, 224, 279, 296, 299, 348, 440, 442, 474, 503, 598, 656, 671, 711, 716, 728, 870, 906, 913, 937]
fp = mol.calcfp()
for m in mols:
f = m.calcfp()
print(f | fp)
0.28 0.3 0.19626168224299065 0.12138728323699421 0.5 0.4666666666666667 0.29577464788732394 0.42857142857142855 0.6176470588235294 0.4117647058823529 0.4772727272727273 0.22826086956521738 0.6363636363636364 1.0
%%html
<div id="chemsim" style="width: 500px"></div>
<script>
var divid = '#chemsim';
jQuery(divid).asker({
id: divid,
question: "What is the Tanimoto coefficient of the bitstrings 1001 and 0011?",
answers: ['0','0.25','0.33','0.5','0.66','0.75','1.0'],
server: "https://bits.csb.pitt.edu/asker.js/example/asker.cgi",
charter: chartmaker})
$(".jp-InputArea .o:contains(html)").closest('.jp-InputArea').hide();
</script>
2D -> 3D¶
mol.make3D() #this makes a reasonable 3D structure
print(mol.atoms[0].coords)
mol.localopt() #this further optimizes the structure
print(mol.atoms[0].coords)
(2.468272539514969, 0.6349880847110817, -0.018730053230247772) (2.46371187360455, 0.6341729709888883, -0.010376048284164541)
sdf = mol.write('sdf')
import py3Dmol
view = py3Dmol.view()
view.addModel(sdf)
view.setStyle({'stick':{}})
view.zoomTo()
view.show()
You appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension:
jupyter labextension install jupyterlab_3dmol
sdf Molecules¶
mols = list(pybel.readfile('sdf','../files/best.sdf'))
len(mols)
10
atom = mols[0].atoms[0]
print(atom.coords)
(-0.5939, -56.8911, 14.3139)
!cat ../files/best.sdf
ZINC78996542
39 44 0 0 0 0 0 0 0 0999 V2000
-0.5939 -56.8911 14.3139 C 0 0 0 0 0 0 0 0 0 0 0 0
2.3154 -57.8883 15.8741 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.3628 -55.5394 14.9296 C 0 0 0 0 0 0 0 0 0 0 0 0
1.0440 -55.7357 15.4805 C 0 0 0 0 0 0 0 0 0 0 0 0
0.3058 -57.7869 14.5684 N 0 0 0 0 0 0 0 0 0 0 0 0
1.2724 -57.1748 15.3144 N 0 0 0 0 0 0 0 0 0 0 0 0
3.1864 -57.3893 16.5881 O 0 0 0 0 0 0 0 0 0 0 0 0
-6.5650 -58.0576 12.9536 C 0 0 0 0 0 0 0 0 0 0 0 0
-6.4112 -58.0403 11.5707 C 0 0 0 0 0 0 0 0 0 0 0 0
-5.4635 -57.8375 13.7859 C 0 0 0 0 0 0 0 0 0 0 0 0
-5.1560 -57.8031 11.0185 C 0 0 0 0 0 0 0 0 0 0 0 0
-4.1883 -57.5962 13.2480 C 0 0 0 0 0 0 0 0 0 0 0 0
-4.0573 -57.5833 11.8565 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.9942 -57.3574 14.1090 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.7971 -57.1312 13.5121 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.6648 -57.1197 12.0139 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.8049 -57.3464 11.2822 N 0 0 0 0 0 0 0 0 0 0 0 0
-0.5742 -56.9136 11.4820 O 0 0 0 0 0 0 0 0 0 0 0 0
-2.7364 -57.3419 10.3001 H 0 0 0 0 0 0 0 0 0 0 0 0
-2.7198 -58.5030 16.2901 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.5010 -56.2171 16.2506 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.7946 -58.5045 17.6831 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.5759 -56.2186 17.6435 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.0729 -57.3592 15.5739 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.2227 -57.3625 18.3596 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.3046 -57.3655 19.8487 C 0 0 0 0 0 0 0 0 0 0 0 0
3.3111 -54.6466 15.3638 C 0 0 0 0 0 0 0 0 0 0 0 0
4.2563 -53.8710 14.6948 C 0 0 0 0 0 0 0 0 0 0 0 0
1.8121 -54.3645 13.5073 C 0 0 0 0 0 0 0 0 0 0 0 0
4.6178 -52.1004 11.5954 C 0 0 0 0 0 0 0 0 0 0 0 0
3.4057 -52.3452 11.0066 C 0 0 0 0 0 0 0 0 0 0 0 0
2.0836 -54.8943 14.7675 C 0 0 0 0 0 0 0 0 0 0 0 0
3.9949 -53.3368 13.4353 C 0 0 0 0 0 0 0 0 0 0 0 0
2.7496 -53.5881 12.8303 C 0 0 0 0 0 0 0 0 0 0 0 0
4.9229 -52.5915 12.8100 N 0 0 0 0 0 0 0 0 0 0 0 0
2.4640 -53.0881 11.6153 N 0 0 0 0 0 0 0 0 0 0 0 0
3.6585 -59.9707 15.9992 C 0 0 0 0 0 0 0 0 0 0 0 0
1.1564 -60.0645 16.2196 C 0 0 0 0 0 0 0 0 0 0 0 0
2.3350 -59.3635 15.5577 C 0 0 0 0 0 0 0 0 0 0 0 0
8 9 1 0 0 0
10 12 1 0 0 0
20 24 1 0 0 0
21 23 1 0 0 0
27 32 1 0 0 0
22 25 1 0 0 0
28 33 1 0 0 0
11 13 1 0 0 0
29 34 1 0 0 0
24 14 1 0 0 0
12 14 1 0 0 0
33 34 1 0 0 0
13 17 1 0 0 0
15 1 1 0 0 0
15 16 1 0 0 0
16 17 1 0 0 0
2 6 1 0 0 0
3 1 1 0 0 0
3 4 1 0 0 0
4 32 1 0 0 0
4 6 1 0 0 0
26 25 1 0 0 0
37 39 1 0 0 0
38 39 1 0 0 0
39 2 1 0 0 0
6 5 1 0 0 0
8 10 2 0 0 0
9 11 2 0 0 0
20 22 2 0 0 0
21 24 2 0 0 0
27 28 2 0 0 0
23 25 2 0 0 0
29 32 2 0 0 0
30 31 2 0 0 0
30 35 2 0 0 0
31 36 2 0 0 0
12 13 2 0 0 0
33 35 2 0 0 0
34 36 2 0 0 0
14 15 2 0 0 0
1 5 2 0 0 0
16 18 2 0 0 0
2 7 2 0 0 0
17 19 1 0 0 0
M END
> <minimizedAffinity>
-7.83433
> <minimizedRMSD>
1.45522
> <molecular weight>
475.372
$$$$
ZINC78996542
39 44 0 0 0 0 0 0 0 0999 V2000
-0.5722 -56.8468 14.3132 C 0 0 0 0 0 0 0 0 0 0 0 0
2.3170 -57.8869 15.8829 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.3244 -55.4995 14.9316 C 0 0 0 0 0 0 0 0 0 0 0 0
1.0775 -55.7161 15.4874 C 0 0 0 0 0 0 0 0 0 0 0 0
0.3140 -57.7556 14.5698 N 0 0 0 0 0 0 0 0 0 0 0 0
1.2862 -57.1582 15.3202 N 0 0 0 0 0 0 0 0 0 0 0 0
3.1923 -57.4012 16.6007 O 0 0 0 0 0 0 0 0 0 0 0 0
-6.5452 -57.9747 12.9290 C 0 0 0 0 0 0 0 0 0 0 0 0
-6.3911 -57.9310 11.5468 C 0 0 0 0 0 0 0 0 0 0 0 0
-5.4434 -57.7729 13.7658 C 0 0 0 0 0 0 0 0 0 0 0 0
-5.1352 -57.6858 10.9997 C 0 0 0 0 0 0 0 0 0 0 0 0
-4.1676 -57.5240 13.2330 C 0 0 0 0 0 0 0 0 0 0 0 0
-4.0362 -57.4846 11.8422 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.9733 -57.3042 14.0987 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.7756 -57.0691 13.5066 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.6429 -57.0290 12.0089 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.7832 -57.2393 11.2727 N 0 0 0 0 0 0 0 0 0 0 0 0
-0.5516 -56.8149 11.4815 O 0 0 0 0 0 0 0 0 0 0 0 0
-2.7144 -57.2159 10.2909 H 0 0 0 0 0 0 0 0 0 0 0 0
-2.7038 -58.4927 16.2574 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.4766 -56.2038 16.2618 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.7789 -58.5209 17.6500 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.5521 -56.2319 17.6545 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.0525 -57.3342 15.5634 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.2031 -57.3905 18.3484 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.2855 -57.4221 19.8372 C 0 0 0 0 0 0 0 0 0 0 0 0
3.3565 -54.6510 15.3845 C 0 0 0 0 0 0 0 0 0 0 0 0
4.3151 -53.8880 14.7200 C 0 0 0 0 0 0 0 0 0 0 0 0
1.8755 -54.3614 13.5148 C 0 0 0 0 0 0 0 0 0 0 0 0
4.7196 -52.1345 11.6161 C 0 0 0 0 0 0 0 0 0 0 0 0
3.5100 -52.3693 11.0185 C 0 0 0 0 0 0 0 0 0 0 0 0
2.1314 -54.8885 14.7794 C 0 0 0 0 0 0 0 0 0 0 0 0
4.0693 -53.3564 13.4560 C 0 0 0 0 0 0 0 0 0 0 0 0
2.8264 -53.5977 12.8421 C 0 0 0 0 0 0 0 0 0 0 0 0
5.0099 -52.6234 12.8352 N 0 0 0 0 0 0 0 0 0 0 0 0
2.5559 -53.0999 11.6229 N 0 0 0 0 0 0 0 0 0 0 0 0
3.6348 -59.9861 16.0001 C 0 0 0 0 0 0 0 0 0 0 0 0
1.1325 -60.0490 16.2302 C 0 0 0 0 0 0 0 0 0 0 0 0
2.3170 -59.3619 15.5646 C 0 0 0 0 0 0 0 0 0 0 0 0
8 9 1 0 0 0
10 12 1 0 0 0
20 24 1 0 0 0
21 23 1 0 0 0
27 32 1 0 0 0
22 25 1 0 0 0
28 33 1 0 0 0
11 13 1 0 0 0
29 34 1 0 0 0
24 14 1 0 0 0
12 14 1 0 0 0
33 34 1 0 0 0
13 17 1 0 0 0
15 1 1 0 0 0
15 16 1 0 0 0
16 17 1 0 0 0
2 6 1 0 0 0
3 1 1 0 0 0
3 4 1 0 0 0
4 32 1 0 0 0
4 6 1 0 0 0
26 25 1 0 0 0
37 39 1 0 0 0
38 39 1 0 0 0
39 2 1 0 0 0
6 5 1 0 0 0
8 10 2 0 0 0
9 11 2 0 0 0
20 22 2 0 0 0
21 24 2 0 0 0
27 28 2 0 0 0
23 25 2 0 0 0
29 32 2 0 0 0
30 31 2 0 0 0
30 35 2 0 0 0
31 36 2 0 0 0
12 13 2 0 0 0
33 35 2 0 0 0
34 36 2 0 0 0
14 15 2 0 0 0
1 5 2 0 0 0
16 18 2 0 0 0
2 7 2 0 0 0
17 19 1 0 0 0
M END
> <minimizedAffinity>
-7.7915
> <minimizedRMSD>
1.18555
> <molecular weight>
475.372
$$$$
ZINC78996534
39 44 0 0 0 0 0 0 0 0999 V2000
-0.6060 -58.4259 14.4308 C 0 0 0 0 0 0 0 0 0 0 0 0
2.2622 -57.0761 15.7885 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.3848 -59.6010 15.3414 C 0 0 0 0 0 0 0 0 0 0 0 0
1.0076 -59.2726 15.8653 C 0 0 0 0 0 0 0 0 0 0 0 0
0.2852 -57.4884 14.4946 N 0 0 0 0 0 0 0 0 0 0 0 0
1.2358 -57.9070 15.3815 N 0 0 0 0 0 0 0 0 0 0 0 0
3.1167 -57.3919 16.6176 O 0 0 0 0 0 0 0 0 0 0 0 0
-6.5490 -57.6468 12.7169 C 0 0 0 0 0 0 0 0 0 0 0 0
-6.3640 -57.9721 11.3766 C 0 0 0 0 0 0 0 0 0 0 0 0
-5.4660 -57.6655 13.6007 C 0 0 0 0 0 0 0 0 0 0 0 0
-5.0959 -58.3170 10.9188 C 0 0 0 0 0 0 0 0 0 0 0 0
-4.1782 -58.0108 13.1580 C 0 0 0 0 0 0 0 0 0 0 0 0
-4.0156 -58.3341 11.8081 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.0030 -58.0395 14.0758 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.7918 -58.3829 13.5703 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.6256 -58.7300 12.1163 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.7497 -58.6825 11.3283 N 0 0 0 0 0 0 0 0 0 0 0 0
-0.5225 -59.0404 11.6676 O 0 0 0 0 0 0 0 0 0 0 0 0
-2.6589 -58.9060 10.3738 H 0 0 0 0 0 0 0 0 0 0 0 0
-3.5198 -58.6843 16.4130 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.8163 -56.4211 15.9438 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.6259 -58.3704 17.7678 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.9225 -56.1072 17.2987 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.1149 -57.7096 15.5009 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.3273 -57.0819 18.2108 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.4433 -56.7463 19.6592 C 0 0 0 0 0 0 0 0 0 0 0 0
2.8030 -59.9702 14.2440 C 0 0 0 0 0 0 0 0 0 0 0 0
3.7737 -60.8749 13.8173 C 0 0 0 0 0 0 0 0 0 0 0 0
2.3083 -61.4219 16.0936 C 0 0 0 0 0 0 0 0 0 0 0 0
5.1631 -64.0341 14.7974 C 0 0 0 0 0 0 0 0 0 0 0 0
4.4366 -64.3052 15.9262 C 0 0 0 0 0 0 0 0 0 0 0 0
2.0669 -60.2450 15.3869 C 0 0 0 0 0 0 0 0 0 0 0 0
4.0225 -62.0537 14.5162 C 0 0 0 0 0 0 0 0 0 0 0 0
3.2759 -62.3324 15.6757 C 0 0 0 0 0 0 0 0 0 0 0 0
4.9642 -62.9109 14.0844 N 0 0 0 0 0 0 0 0 0 0 0 0
3.4901 -63.4609 16.3744 N 0 0 0 0 0 0 0 0 0 0 0 0
1.2882 -54.7944 15.8358 C 0 0 0 0 0 0 0 0 0 0 0 0
3.6925 -55.1298 15.1839 C 0 0 0 0 0 0 0 0 0 0 0 0
2.2860 -55.7119 15.1441 C 0 0 0 0 0 0 0 0 0 0 0 0
8 9 1 0 0 0
10 12 1 0 0 0
20 24 1 0 0 0
21 23 1 0 0 0
27 32 1 0 0 0
22 25 1 0 0 0
28 33 1 0 0 0
11 13 1 0 0 0
29 34 1 0 0 0
24 14 1 0 0 0
12 14 1 0 0 0
33 34 1 0 0 0
13 17 1 0 0 0
15 1 1 0 0 0
15 16 1 0 0 0
16 17 1 0 0 0
2 6 1 0 0 0
3 1 1 0 0 0
3 4 1 0 0 0
4 32 1 0 0 0
4 6 1 0 0 0
26 25 1 0 0 0
37 39 1 0 0 0
38 39 1 0 0 0
39 2 1 0 0 0
6 5 1 0 0 0
8 10 2 0 0 0
9 11 2 0 0 0
20 22 2 0 0 0
21 24 2 0 0 0
27 28 2 0 0 0
23 25 2 0 0 0
29 32 2 0 0 0
30 31 2 0 0 0
30 35 2 0 0 0
31 36 2 0 0 0
12 13 2 0 0 0
33 35 2 0 0 0
34 36 2 0 0 0
14 15 2 0 0 0
1 5 2 0 0 0
16 18 2 0 0 0
2 7 2 0 0 0
17 19 1 0 0 0
M END
> <minimizedAffinity>
-7.60183
> <minimizedRMSD>
2.26383
> <molecular weight>
475.372
$$$$
ZINC78996542
39 44 0 0 0 0 0 0 0 0999 V2000
-1.1562 -57.7105 14.6555 C 0 0 0 0 0 0 0 0 0 0 0 0
2.0859 -57.6546 15.8298 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.1390 -56.2126 14.7797 C 0 0 0 0 0 0 0 0 0 0 0 0
0.3392 -55.9873 15.0720 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.0584 -58.3149 14.9816 N 0 0 0 0 0 0 0 0 0 0 0 0
0.8469 -57.3439 15.3020 N 0 0 0 0 0 0 0 0 0 0 0 0
2.9188 -56.8145 16.1731 O 0 0 0 0 0 0 0 0 0 0 0 0
-6.8917 -60.1405 14.9063 C 0 0 0 0 0 0 0 0 0 0 0 0
-6.9219 -60.5932 13.5908 C 0 0 0 0 0 0 0 0 0 0 0 0
-5.7606 -59.4813 15.3968 C 0 0 0 0 0 0 0 0 0 0 0 0
-5.8212 -60.3875 12.7645 C 0 0 0 0 0 0 0 0 0 0 0 0
-4.6396 -59.2637 14.5786 C 0 0 0 0 0 0 0 0 0 0 0 0
-4.6919 -59.7269 13.2610 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.4188 -58.5636 15.0722 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.3780 -58.3922 14.2190 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.4425 -58.8944 12.8026 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.5968 -59.5297 12.4148 N 0 0 0 0 0 0 0 0 0 0 0 0
-1.4927 -58.7341 12.0364 O 0 0 0 0 0 0 0 0 0 0 0 0
-3.6562 -59.8644 11.4908 H 0 0 0 0 0 0 0 0 0 0 0 0
-2.7756 -58.8669 17.4469 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.7367 -56.7639 16.7466 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.6706 -58.3845 18.7515 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.6320 -56.2815 18.0513 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.3085 -58.0565 16.4443 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.0988 -57.0919 19.0536 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.9885 -56.5777 20.4491 C 0 0 0 0 0 0 0 0 0 0 0 0
0.6434 -55.4303 12.6355 C 0 0 0 0 0 0 0 0 0 0 0 0
1.3219 -54.7691 11.6131 C 0 0 0 0 0 0 0 0 0 0 0 0
2.1612 -54.4616 14.2263 C 0 0 0 0 0 0 0 0 0 0 0 0
4.1096 -52.5500 11.1925 C 0 0 0 0 0 0 0 0 0 0 0 0
4.5256 -52.3975 12.4883 C 0 0 0 0 0 0 0 0 0 0 0 0
1.0650 -55.2760 13.9477 C 0 0 0 0 0 0 0 0 0 0 0 0
2.4186 -53.9532 11.8808 C 0 0 0 0 0 0 0 0 0 0 0 0
2.8462 -53.7965 13.2121 C 0 0 0 0 0 0 0 0 0 0 0 0
3.0567 -53.3258 10.8773 N 0 0 0 0 0 0 0 0 0 0 0 0
3.9009 -53.0163 13.5062 N 0 0 0 0 0 0 0 0 0 0 0 0
2.4158 -59.7838 14.5992 C 0 0 0 0 0 0 0 0 0 0 0 0
3.6872 -59.3395 16.7216 C 0 0 0 0 0 0 0 0 0 0 0 0
2.3789 -59.1280 15.9719 C 0 0 0 0 0 0 0 0 0 0 0 0
8 9 1 0 0 0
10 12 1 0 0 0
20 24 1 0 0 0
21 23 1 0 0 0
27 32 1 0 0 0
22 25 1 0 0 0
28 33 1 0 0 0
11 13 1 0 0 0
29 34 1 0 0 0
24 14 1 0 0 0
12 14 1 0 0 0
33 34 1 0 0 0
13 17 1 0 0 0
15 1 1 0 0 0
15 16 1 0 0 0
16 17 1 0 0 0
2 6 1 0 0 0
3 1 1 0 0 0
3 4 1 0 0 0
4 32 1 0 0 0
4 6 1 0 0 0
26 25 1 0 0 0
37 39 1 0 0 0
38 39 1 0 0 0
39 2 1 0 0 0
6 5 1 0 0 0
8 10 2 0 0 0
9 11 2 0 0 0
20 22 2 0 0 0
21 24 2 0 0 0
27 28 2 0 0 0
23 25 2 0 0 0
29 32 2 0 0 0
30 31 2 0 0 0
30 35 2 0 0 0
31 36 2 0 0 0
12 13 2 0 0 0
33 35 2 0 0 0
34 36 2 0 0 0
14 15 2 0 0 0
1 5 2 0 0 0
16 18 2 0 0 0
2 7 2 0 0 0
17 19 1 0 0 0
M END
> <minimizedAffinity>
-7.58798
> <minimizedRMSD>
1.876
> <molecular weight>
475.372
$$$$
ZINC35448294
33 38 0 0 0 0 0 0 0 0999 V2000
6.2193 -51.5392 13.7822 C 0 0 0 0 0 0 0 0 0 0 0 0
6.5893 -51.9773 15.0518 C 0 0 0 0 0 0 0 0 0 0 0 0
5.1156 -52.0958 13.1259 C 0 0 0 0 0 0 0 0 0 0 0 0
5.8703 -52.9821 15.7052 C 0 0 0 0 0 0 0 0 0 0 0 0
4.3763 -53.1129 13.7626 C 0 0 0 0 0 0 0 0 0 0 0 0
3.2347 -53.8835 13.4110 C 0 0 0 0 0 0 0 0 0 0 0 0
4.7696 -53.5311 15.0379 C 0 0 0 0 0 0 0 0 0 0 0 0
2.9678 -54.7425 14.4550 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.2954 -56.1404 13.1784 C 0 0 0 0 0 0 0 0 0 0 0 0
2.3853 -53.8636 12.1948 C 0 0 0 0 0 0 0 0 0 0 0 0
1.6865 -55.2195 12.0267 C 0 0 0 0 0 0 0 0 0 0 0 0
1.8500 -55.7376 14.5366 C 0 0 0 0 0 0 0 0 0 0 0 0
3.8912 -54.5146 15.4387 N 0 0 0 0 0 0 0 0 0 0 0 0
1.0317 -55.6866 13.2732 N 0 0 0 0 0 0 0 0 0 0 0 0
-0.9662 -56.1848 12.1474 O 0 0 0 0 0 0 0 0 0 0 0 0
3.9233 -54.9834 16.3038 H 0 0 0 0 0 0 0 0 0 0 0 0
-2.9363 -58.4538 18.1389 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.3508 -57.1319 18.2899 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.1432 -58.8360 17.0510 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.9875 -56.1521 17.3616 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.9885 -57.8949 14.9095 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.7643 -57.8655 16.1021 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.1944 -56.5486 16.2790 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.9633 -56.6191 14.3950 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.6932 -55.8143 15.2279 N 0 0 0 0 0 0 0 0 0 0 0 0
-1.8386 -54.8497 15.0945 H 0 0 0 0 0 0 0 0 0 0 0 0
2.4658 -57.5272 16.2097 C 0 0 0 0 0 0 0 0 0 0 0 0
2.6382 -58.0380 13.8546 C 0 0 0 0 0 0 0 0 0 0 0 0
2.9083 -58.8130 16.5209 C 0 0 0 0 0 0 0 0 0 0 0 0
3.0807 -59.3238 14.1659 C 0 0 0 0 0 0 0 0 0 0 0 0
2.3307 -57.1398 14.8765 C 0 0 0 0 0 0 0 0 0 0 0 0
3.2157 -59.7112 15.4991 C 0 0 0 0 0 0 0 0 0 0 0 0
3.7617 -61.2970 15.8834 Cl 0 0 0 0 0 0 0 0 0 0 0 0
17 18 1 0 0 0
1 2 1 0 0 0
19 22 1 0 0 0
3 5 1 0 0 0
27 31 1 0 0 0
28 30 1 0 0 0
20 23 1 0 0 0
4 7 1 0 0 0
29 32 1 0 0 0
21 22 1 0 0 0
5 6 1 0 0 0
23 25 1 0 0 0
7 13 1 0 0 0
32 33 1 0 0 0
24 9 1 0 0 0
24 25 1 0 0 0
8 13 1 0 0 0
9 14 1 0 0 0
10 6 1 0 0 0
10 11 1 0 0 0
11 14 1 0 0 0
12 31 1 0 0 0
12 8 1 0 0 0
12 14 1 0 0 0
17 19 2 0 0 0
1 3 2 0 0 0
18 20 2 0 0 0
2 4 2 0 0 0
27 29 2 0 0 0
28 31 2 0 0 0
30 32 2 0 0 0
21 24 2 0 0 0
22 23 2 0 0 0
5 7 2 0 0 0
6 8 2 0 0 0
9 15 2 0 0 0
25 26 1 0 0 0
13 16 1 0 0 0
M END
> <minimizedAffinity>
-7.52352
> <minimizedRMSD>
6.72818
> <molecular weight>
407.767
$$$$
ZINC72314638
34 38 0 0 0 0 0 0 0 0999 V2000
-6.9192 -60.0249 14.8267 C 0 0 0 0 0 0 0 0 0 0 0 0
-6.9224 -60.5532 13.5394 C 0 0 0 0 0 0 0 0 0 0 0 0
-5.7591 -59.4408 15.3438 C 0 0 0 0 0 0 0 0 0 0 0 0
-5.7655 -60.4981 12.7677 C 0 0 0 0 0 0 0 0 0 0 0 0
-4.5814 -59.3754 14.5808 C 0 0 0 0 0 0 0 0 0 0 0 0
-4.6074 -59.9121 13.2905 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.3284 -58.7584 15.1036 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.2330 -58.7339 14.3037 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.2694 -59.3144 12.9167 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.4559 -59.8663 12.4993 N 0 0 0 0 0 0 0 0 0 0 0 0
-1.2700 -59.2872 12.1989 O 0 0 0 0 0 0 0 0 0 0 0 0
-3.4977 -60.2504 11.5937 H 0 0 0 0 0 0 0 0 0 0 0 0
-0.9776 -58.1386 14.7709 C 0 0 0 0 0 0 0 0 0 0 0 0
1.1138 -55.4726 15.4117 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0692 -59.0069 15.4111 C 0 0 0 0 0 0 0 0 0 0 0 0
1.2024 -57.9981 15.5499 C 0 0 0 0 0 0 0 0 0 0 0 0
2.4884 -55.4636 16.0338 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.6963 -56.8766 14.7008 N 0 0 0 0 0 0 0 0 0 0 0 0
0.5616 -56.7232 15.2099 N 0 0 0 0 0 0 0 0 0 0 0 0
0.5532 -54.4163 15.1165 O 0 0 0 0 0 0 0 0 0 0 0 0
3.3449 -58.4107 12.4428 C 0 0 0 0 0 0 0 0 0 0 0 0
4.5462 -58.8702 12.9824 C 0 0 0 0 0 0 0 0 0 0 0 0
2.2658 -58.1304 13.2810 C 0 0 0 0 0 0 0 0 0 0 0 0
4.6683 -59.0498 14.3602 C 0 0 0 0 0 0 0 0 0 0 0 0
2.3878 -58.3101 14.6587 C 0 0 0 0 0 0 0 0 0 0 0 0
3.5892 -58.7698 15.1985 C 0 0 0 0 0 0 0 0 0 0 0 0
3.7434 -58.9704 16.6705 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.8700 -58.9787 17.5299 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.5363 -56.8305 16.6476 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.7893 -58.4284 18.8090 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.4557 -56.2801 17.9268 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.2435 -58.1798 16.4491 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.0821 -57.0791 19.0075 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.9979 -56.4920 20.3757 C 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0 0 0
21 22 1 0 0 0
3 5 1 0 0 0
28 32 1 0 0 0
29 31 1 0 0 0
23 25 1 0 0 0
24 26 1 0 0 0
30 33 1 0 0 0
4 6 1 0 0 0
32 7 1 0 0 0
5 7 1 0 0 0
6 10 1 0 0 0
8 13 1 0 0 0
8 9 1 0 0 0
9 10 1 0 0 0
14 19 1 0 0 0
15 13 1 0 0 0
15 16 1 0 0 0
16 25 1 0 0 0
16 19 1 0 0 0
34 33 1 0 0 0
27 26 1 0 0 0
17 14 1 0 0 0
19 18 1 0 0 0
1 3 2 0 0 0
21 23 2 0 0 0
22 24 2 0 0 0
2 4 2 0 0 0
28 30 2 0 0 0
29 32 2 0 0 0
31 33 2 0 0 0
5 6 2 0 0 0
25 26 2 0 0 0
7 8 2 0 0 0
13 18 2 0 0 0
9 11 2 0 0 0
14 20 2 0 0 0
10 12 1 0 0 0
M END
> <minimizedAffinity>
-7.51168
> <minimizedRMSD>
1.81673
> <molecular weight>
411.326
$$$$
ZINC72314638
34 38 0 0 0 0 0 0 0 0999 V2000
-6.9192 -60.0250 14.8266 C 0 0 0 0 0 0 0 0 0 0 0 0
-6.9223 -60.5532 13.5393 C 0 0 0 0 0 0 0 0 0 0 0 0
-5.7590 -59.4409 15.3438 C 0 0 0 0 0 0 0 0 0 0 0 0
-5.7655 -60.4980 12.7677 C 0 0 0 0 0 0 0 0 0 0 0 0
-4.5813 -59.3753 14.5807 C 0 0 0 0 0 0 0 0 0 0 0 0
-4.6073 -59.9120 13.2905 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.3283 -58.7583 15.1036 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.2330 -58.7336 14.3037 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.2693 -59.3140 12.9166 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.4559 -59.8659 12.4992 N 0 0 0 0 0 0 0 0 0 0 0 0
-1.2700 -59.2867 12.1988 O 0 0 0 0 0 0 0 0 0 0 0 0
-3.4977 -60.2499 11.5936 H 0 0 0 0 0 0 0 0 0 0 0 0
-0.9776 -58.1384 14.7708 C 0 0 0 0 0 0 0 0 0 0 0 0
1.1137 -55.4723 15.4114 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0691 -59.0065 15.4111 C 0 0 0 0 0 0 0 0 0 0 0 0
1.2024 -57.9977 15.5499 C 0 0 0 0 0 0 0 0 0 0 0 0
2.4883 -55.4632 16.0337 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.6963 -56.8762 14.7006 N 0 0 0 0 0 0 0 0 0 0 0 0
0.5616 -56.7229 15.2098 N 0 0 0 0 0 0 0 0 0 0 0 0
0.5532 -54.4160 15.1161 O 0 0 0 0 0 0 0 0 0 0 0 0
3.3452 -58.4103 12.4429 C 0 0 0 0 0 0 0 0 0 0 0 0
4.5464 -58.8703 12.9827 C 0 0 0 0 0 0 0 0 0 0 0 0
2.2660 -58.1300 13.2811 C 0 0 0 0 0 0 0 0 0 0 0 0
4.6682 -59.0500 14.3606 C 0 0 0 0 0 0 0 0 0 0 0 0
2.3879 -58.3098 14.6589 C 0 0 0 0 0 0 0 0 0 0 0 0
3.5890 -58.7698 15.1986 C 0 0 0 0 0 0 0 0 0 0 0 0
3.7431 -58.9706 16.6707 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.8702 -58.9787 17.5299 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.5360 -56.8304 16.6476 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.7895 -58.4286 18.8091 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.4554 -56.2801 17.9268 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.2434 -58.1797 16.4491 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.0819 -57.0792 19.0075 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.9978 -56.4922 20.3758 C 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0 0 0
21 22 1 0 0 0
3 5 1 0 0 0
28 32 1 0 0 0
29 31 1 0 0 0
23 25 1 0 0 0
24 26 1 0 0 0
30 33 1 0 0 0
4 6 1 0 0 0
32 7 1 0 0 0
5 7 1 0 0 0
6 10 1 0 0 0
8 13 1 0 0 0
8 9 1 0 0 0
9 10 1 0 0 0
14 19 1 0 0 0
15 13 1 0 0 0
15 16 1 0 0 0
16 25 1 0 0 0
16 19 1 0 0 0
34 33 1 0 0 0
27 26 1 0 0 0
17 14 1 0 0 0
19 18 1 0 0 0
1 3 2 0 0 0
21 23 2 0 0 0
22 24 2 0 0 0
2 4 2 0 0 0
28 30 2 0 0 0
29 32 2 0 0 0
31 33 2 0 0 0
5 6 2 0 0 0
25 26 2 0 0 0
7 8 2 0 0 0
13 18 2 0 0 0
9 11 2 0 0 0
14 20 2 0 0 0
10 12 1 0 0 0
M END
> <minimizedAffinity>
-7.51156
> <minimizedRMSD>
2.07052
> <molecular weight>
411.326
$$$$
ZINC39912421
35 39 0 0 0 0 0 0 0 0999 V2000
-2.8637 -58.0485 14.3831 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.6591 -57.4567 14.0111 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.7718 -57.6110 13.4624 C 0 0 0 0 0 0 0 0 0 0 0 0
-5.0742 -58.1435 13.6832 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.4965 -58.9601 15.3604 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.8048 -56.6783 12.9233 N 0 0 0 0 0 0 0 0 0 0 0 0
-3.1200 -56.7955 12.6090 N 0 0 0 0 0 0 0 0 0 0 0 0
-4.8956 -58.9553 14.8275 N 0 0 0 0 0 0 0 0 0 0 0 0
-6.0866 -57.9453 13.0303 O 0 0 0 0 0 0 0 0 0 0 0 0
-3.5472 -56.3394 11.8484 H 0 0 0 0 0 0 0 0 0 0 0 0
0.0881 -58.8830 14.9464 C 0 0 0 0 0 0 0 0 0 0 0 0
2.1072 -57.9326 15.8719 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.3763 -57.6028 14.6449 C 0 0 0 0 0 0 0 0 0 0 0 0
1.3300 -59.0479 15.5599 C 0 0 0 0 0 0 0 0 0 0 0 0
1.6430 -56.6523 15.5704 C 0 0 0 0 0 0 0 0 0 0 0 0
0.4011 -56.4874 14.9569 C 0 0 0 0 0 0 0 0 0 0 0 0
1.8249 -60.4168 15.8846 C 0 0 0 0 0 0 0 0 0 0 0 0
2.4900 -55.4712 15.9138 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.0441 -55.2299 14.6663 O 0 0 0 0 0 0 0 0 0 0 0 0
0.4836 -54.4853 14.8788 H 0 0 0 0 0 0 0 0 0 0 0 0
-2.9761 -58.9905 17.8000 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.6595 -56.9124 16.7746 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.8625 -58.3455 19.0315 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.5458 -56.2672 18.0061 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.3746 -58.2738 16.6715 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.1474 -56.9839 19.1346 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.0409 -56.3650 20.3177 F 0 0 0 0 0 0 0 0 0 0 0 0
-5.9715 -59.7155 15.4305 C 0 0 0 0 0 0 0 0 0 0 0 0
-8.5076 -61.3824 13.1820 C 0 0 0 0 0 0 0 0 0 0 0 0
-7.7831 -62.4617 12.6762 C 0 0 0 0 0 0 0 0 0 0 0 0
-7.9064 -60.4967 14.0761 C 0 0 0 0 0 0 0 0 0 0 0 0
-6.4575 -62.6553 13.0647 C 0 0 0 0 0 0 0 0 0 0 0 0
-6.5808 -60.6902 14.4647 C 0 0 0 0 0 0 0 0 0 0 0 0
-5.8563 -61.7695 13.9589 C 0 0 0 0 0 0 0 0 0 0 0 0
-4.2144 -62.0410 14.4164 Cl 0 0 0 0 0 0 0 0 0 0 0 0
29 30 1 0 0 0
21 25 1 0 0 0
22 24 1 0 0 0
31 33 1 0 0 0
23 26 1 0 0 0
32 34 1 0 0 0
11 13 1 0 0 0
12 14 1 0 0 0
13 2 1 0 0 0
1 2 1 0 0 0
15 16 1 0 0 0
16 19 1 0 0 0
26 27 1 0 0 0
34 35 1 0 0 0
3 4 1 0 0 0
3 7 1 0 0 0
4 8 1 0 0 0
5 25 1 0 0 0
5 1 1 0 0 0
5 8 1 0 0 0
17 14 1 0 0 0
18 15 1 0 0 0
28 33 1 0 0 0
28 8 1 0 0 0
7 6 1 0 0 0
29 31 2 0 0 0
30 32 2 0 0 0
21 23 2 0 0 0
22 25 2 0 0 0
24 26 2 0 0 0
11 14 2 0 0 0
12 15 2 0 0 0
13 16 2 0 0 0
1 3 2 0 0 0
33 34 2 0 0 0
2 6 2 0 0 0
4 9 2 0 0 0
7 10 1 0 0 0
19 20 1 0 0 0
M END
> <minimizedAffinity>
-7.49363
> <minimizedRMSD>
1.94002
> <molecular weight>
442.764
$$$$
ZINC39912421
35 39 0 0 0 0 0 0 0 0999 V2000
-2.8637 -58.0484 14.3832 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.6591 -57.4566 14.0111 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.7718 -57.6110 13.4624 C 0 0 0 0 0 0 0 0 0 0 0 0
-5.0743 -58.1434 13.6832 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.4964 -58.9600 15.3605 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.8048 -56.6783 12.9233 N 0 0 0 0 0 0 0 0 0 0 0 0
-3.1200 -56.7954 12.6090 N 0 0 0 0 0 0 0 0 0 0 0 0
-4.8956 -58.9552 14.8276 N 0 0 0 0 0 0 0 0 0 0 0 0
-6.0865 -57.9453 13.0303 O 0 0 0 0 0 0 0 0 0 0 0 0
-3.5473 -56.3392 11.8484 H 0 0 0 0 0 0 0 0 0 0 0 0
0.0881 -58.8830 14.9464 C 0 0 0 0 0 0 0 0 0 0 0 0
2.1074 -57.9325 15.8720 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.3763 -57.6028 14.6449 C 0 0 0 0 0 0 0 0 0 0 0 0
1.3299 -59.0479 15.5600 C 0 0 0 0 0 0 0 0 0 0 0 0
1.6428 -56.6523 15.5704 C 0 0 0 0 0 0 0 0 0 0 0 0
0.4011 -56.4874 14.9569 C 0 0 0 0 0 0 0 0 0 0 0 0
1.8249 -60.4168 15.8846 C 0 0 0 0 0 0 0 0 0 0 0 0
2.4900 -55.4712 15.9137 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.0441 -55.2299 14.6664 O 0 0 0 0 0 0 0 0 0 0 0 0
0.4836 -54.4854 14.8789 H 0 0 0 0 0 0 0 0 0 0 0 0
-2.9763 -58.9904 17.8002 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.6592 -56.9122 16.7746 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.8627 -58.3454 19.0317 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.5455 -56.2671 18.0061 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.3746 -58.2738 16.6716 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.1473 -56.9838 19.1347 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.0408 -56.3649 20.3178 F 0 0 0 0 0 0 0 0 0 0 0 0
-5.9714 -59.7154 15.4306 C 0 0 0 0 0 0 0 0 0 0 0 0
-8.5075 -61.3823 13.1820 C 0 0 0 0 0 0 0 0 0 0 0 0
-7.7831 -62.4617 12.6762 C 0 0 0 0 0 0 0 0 0 0 0 0
-7.9063 -60.4966 14.0762 C 0 0 0 0 0 0 0 0 0 0 0 0
-6.4575 -62.6552 13.0648 C 0 0 0 0 0 0 0 0 0 0 0 0
-6.5808 -60.6901 14.4648 C 0 0 0 0 0 0 0 0 0 0 0 0
-5.8563 -61.7695 13.9591 C 0 0 0 0 0 0 0 0 0 0 0 0
-4.2143 -62.0411 14.4166 Cl 0 0 0 0 0 0 0 0 0 0 0 0
29 30 1 0 0 0
21 25 1 0 0 0
22 24 1 0 0 0
31 33 1 0 0 0
23 26 1 0 0 0
32 34 1 0 0 0
11 13 1 0 0 0
12 14 1 0 0 0
13 2 1 0 0 0
1 2 1 0 0 0
15 16 1 0 0 0
16 19 1 0 0 0
26 27 1 0 0 0
34 35 1 0 0 0
3 4 1 0 0 0
3 7 1 0 0 0
4 8 1 0 0 0
5 25 1 0 0 0
5 1 1 0 0 0
5 8 1 0 0 0
17 14 1 0 0 0
18 15 1 0 0 0
28 33 1 0 0 0
28 8 1 0 0 0
7 6 1 0 0 0
29 31 2 0 0 0
30 32 2 0 0 0
21 23 2 0 0 0
22 25 2 0 0 0
24 26 2 0 0 0
11 14 2 0 0 0
12 15 2 0 0 0
13 16 2 0 0 0
1 3 2 0 0 0
33 34 2 0 0 0
2 6 2 0 0 0
4 9 2 0 0 0
7 10 1 0 0 0
19 20 1 0 0 0
M END
> <minimizedAffinity>
-7.49359
> <minimizedRMSD>
2.12367
> <molecular weight>
442.764
$$$$
ZINC39912344
35 39 0 0 0 0 0 0 0 0999 V2000
-2.9655 -58.0579 14.4075 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.7487 -57.5053 14.0153 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.8718 -57.6016 13.4941 C 0 0 0 0 0 0 0 0 0 0 0 0
-5.1866 -58.0933 13.7357 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.6126 -58.9419 15.4006 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.8851 -56.7323 12.9225 N 0 0 0 0 0 0 0 0 0 0 0 0
-3.2070 -56.8131 12.6256 N 0 0 0 0 0 0 0 0 0 0 0 0
-5.0176 -58.9002 14.8849 N 0 0 0 0 0 0 0 0 0 0 0 0
-6.2006 -57.8708 13.0937 O 0 0 0 0 0 0 0 0 0 0 0 0
-3.6301 -56.3508 11.8663 H 0 0 0 0 0 0 0 0 0 0 0 0
-0.0185 -58.9767 14.9117 C 0 0 0 0 0 0 0 0 0 0 0 0
2.0258 -58.0767 15.8325 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.4630 -57.6840 14.6346 C 0 0 0 0 0 0 0 0 0 0 0 0
1.2258 -59.1731 15.5107 C 0 0 0 0 0 0 0 0 0 0 0 0
1.5813 -56.7838 15.5554 C 0 0 0 0 0 0 0 0 0 0 0 0
0.3369 -56.5875 14.9564 C 0 0 0 0 0 0 0 0 0 0 0 0
1.6995 -60.5554 15.8092 C 0 0 0 0 0 0 0 0 0 0 0 0
2.4524 -55.6234 15.9088 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.0884 -55.3180 14.6896 O 0 0 0 0 0 0 0 0 0 0 0 0
0.4544 -54.5863 14.9086 H 0 0 0 0 0 0 0 0 0 0 0 0
-3.0661 -58.9675 17.8345 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.6938 -56.8776 16.7976 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.9180 -58.3157 19.0588 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.5456 -56.2257 18.0218 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.4539 -58.2484 16.7039 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.1577 -56.9448 19.1524 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.0181 -56.3191 20.3285 F 0 0 0 0 0 0 0 0 0 0 0 0
-6.1077 -59.6230 15.5080 C 0 0 0 0 0 0 0 0 0 0 0 0
-8.0602 -62.0396 13.3356 C 0 0 0 0 0 0 0 0 0 0 0 0
-5.9663 -63.0767 13.9497 C 0 0 0 0 0 0 0 0 0 0 0 0
-7.7088 -60.9008 14.0603 C 0 0 0 0 0 0 0 0 0 0 0 0
-5.6148 -61.9379 14.6743 C 0 0 0 0 0 0 0 0 0 0 0 0
-7.1891 -63.1276 13.2804 C 0 0 0 0 0 0 0 0 0 0 0 0
-6.4861 -60.8499 14.7295 C 0 0 0 0 0 0 0 0 0 0 0 0
-7.5641 -64.3456 12.5058 C 0 0 0 0 0 0 0 0 0 0 0 0
21 25 1 0 0 0
22 24 1 0 0 0
29 33 1 0 0 0
30 32 1 0 0 0
31 34 1 0 0 0
23 26 1 0 0 0
11 13 1 0 0 0
12 14 1 0 0 0
13 2 1 0 0 0
1 2 1 0 0 0
15 16 1 0 0 0
16 19 1 0 0 0
26 27 1 0 0 0
3 4 1 0 0 0
3 7 1 0 0 0
4 8 1 0 0 0
5 25 1 0 0 0
5 1 1 0 0 0
5 8 1 0 0 0
35 33 1 0 0 0
17 14 1 0 0 0
18 15 1 0 0 0
28 34 1 0 0 0
28 8 1 0 0 0
7 6 1 0 0 0
21 23 2 0 0 0
22 25 2 0 0 0
29 31 2 0 0 0
30 33 2 0 0 0
32 34 2 0 0 0
24 26 2 0 0 0
11 14 2 0 0 0
12 15 2 0 0 0
13 16 2 0 0 0
1 3 2 0 0 0
2 6 2 0 0 0
4 9 2 0 0 0
7 10 1 0 0 0
19 20 1 0 0 0
M END
> <minimizedAffinity>
-7.4906
> <minimizedRMSD>
1.79745
> <molecular weight>
419.322
$$$$
sdf files can have arbitrary data embedded in them:
M END
> <minimizedAffinity>
-7.83433
> <minimizedRMSD>
1.45522
> <molecular weight>
475.372
$$$$mols[0].data
{'MOL Chiral Flag': '0', 'minimizedAffinity': '-7.83433', 'minimizedRMSD': '1.45522', 'molecular weight': '475.372', 'OpenBabel Symmetry Classes': '27 24 14 23 12 26 2 6 7 16 15 31 35 32 29 34 30 4 5 13 13 11 11 28 21 1 10 17 20 9 8 25 36 33 18 19 3 3 22'}
Beyond Pybel¶
Recall that Pybel is a python-native wrapper around the OpenBabel SWIG bindings. The underlying OpenBabel objects are always accessible if you need to use the additional functionality provided by OpenBabel (this may be necessary if you modifying or creating molecule objects).
obmol = mol.OBMol
vec = obmol.Center(0)
print(vec)
<openbabel.openbabel.vector3; proxy of <Swig Object of type 'OpenBabel::vector3 *' at 0x7f6af63aa610> >
print(vec.GetX(),vec.GetY(),vec.GetZ())
0.7085902947538542 -0.016861359909934506 -0.0005521894831451903
dir(obmol)
['AddAtom', 'AddBond', 'AddConformer', 'AddHydrogens', 'AddNewHydrogens', 'AddNonPolarHydrogens', 'AddPolarHydrogens', 'AddResidue', 'Align', 'AreInSameRing', 'AssignSpinMultiplicity', 'AssignTotalChargeToAtoms', 'AutomaticFormalCharge', 'AutomaticPartialCharge', 'BeginAtom', 'BeginAtoms', 'BeginBond', 'BeginBonds', 'BeginConformer', 'BeginData', 'BeginInternalCoord', 'BeginModify', 'BeginResidue', 'BeginResidues', 'CBeginAtoms', 'CEndAtoms', 'Center', 'ClassDescription', 'Clear', 'CloneData', 'ConnectTheDots', 'ContigFragList', 'ConvertDativeBonds', 'ConvertZeroBonds', 'CopyConformer', 'CopySubstructure', 'CorrectForPH', 'DataSize', 'DecrementMod', 'DeleteAtom', 'DeleteBond', 'DeleteConformer', 'DeleteData', 'DeleteHydrogen', 'DeleteHydrogens', 'DeleteNonPolarHydrogens', 'DeletePolarHydrogens', 'DeleteResidue', 'DestroyAtom', 'DestroyBond', 'DestroyResidue', 'DoTransformations', 'Empty', 'EndAtom', 'EndAtoms', 'EndBond', 'EndBonds', 'EndData', 'EndModify', 'EndResidue', 'EndResidues', 'FindAngles', 'FindChildren', 'FindLSSR', 'FindLargestFragment', 'FindRingAtomsAndBonds', 'FindSSSR', 'FindTorsions', 'GetAllData', 'GetAngle', 'GetAtom', 'GetAtomById', 'GetBond', 'GetBondById', 'GetConformer', 'GetConformers', 'GetCoordinates', 'GetData', 'GetDimension', 'GetEnergies', 'GetEnergy', 'GetExactMass', 'GetFirstAtom', 'GetFlags', 'GetFormula', 'GetGIDVector', 'GetGIVector', 'GetGTDVector', 'GetInternalCoord', 'GetLSSR', 'GetMod', 'GetMolWt', 'GetNextFragment', 'GetResidue', 'GetSSSR', 'GetSpacedFormula', 'GetTitle', 'GetTorsion', 'GetTotalCharge', 'GetTotalSpinMultiplicity', 'Has2D', 'Has3D', 'HasAromaticPerceived', 'HasAtomTypesPerceived', 'HasChainsPerceived', 'HasChiralityPerceived', 'HasClosureBondsPerceived', 'HasData', 'HasFlag', 'HasHybridizationPerceived', 'HasHydrogensAdded', 'HasLSSRPerceived', 'HasNonZeroCoords', 'HasPartialChargesPerceived', 'HasRingAtomsAndBondsPerceived', 'HasRingTypesPerceived', 'HasSSSRPerceived', 'HasSpinMultiplicityAssigned', 'IncrementMod', 'InsertAtom', 'IsCorrectedForPH', 'IsPeriodic', 'IsReaction', 'MakeDativeBonds', 'NewAtom', 'NewBond', 'NewResidue', 'NextConformer', 'NextInternalCoord', 'NumAtoms', 'NumBonds', 'NumConformers', 'NumHvyAtoms', 'NumResidues', 'NumRotors', 'PerceiveBondOrders', 'RenumberAtoms', 'ReserveAtoms', 'Rotate', 'Separate', 'SetAromaticPerceived', 'SetAtomTypesPerceived', 'SetAutomaticFormalCharge', 'SetAutomaticPartialCharge', 'SetChainsPerceived', 'SetChiralityPerceived', 'SetClosureBondsPerceived', 'SetConformer', 'SetConformers', 'SetCoordinates', 'SetCorrectedForPH', 'SetData', 'SetDimension', 'SetEnergies', 'SetEnergy', 'SetFlag', 'SetFlags', 'SetFormula', 'SetHybridizationPerceived', 'SetHydrogensAdded', 'SetInternalCoord', 'SetIsPatternStructure', 'SetIsReaction', 'SetLSSRPerceived', 'SetPartialChargesPerceived', 'SetPeriodicMol', 'SetRingAtomsAndBondsPerceived', 'SetRingTypesPerceived', 'SetSSSRPerceived', 'SetSpinMultiplicityAssigned', 'SetTitle', 'SetTorsion', 'SetTotalCharge', 'SetTotalSpinMultiplicity', 'StripSalts', 'ToInertialFrame', 'Translate', 'UnsetFlag', '__class__', '__delattr__', '__dict__', '__dir__', '__doc__', '__eq__', '__format__', '__ge__', '__getattribute__', '__gt__', '__hash__', '__iadd__', '__init__', '__init_subclass__', '__le__', '__lt__', '__module__', '__ne__', '__new__', '__reduce__', '__reduce_ex__', '__repr__', '__setattr__', '__sizeof__', '__str__', '__subclasshook__', '__swig_destroy__', '__weakref__', 'this', 'thisown']
Project: Dimensionality Reduced Molecules¶
Given a SMILES file where the molecules names are property (e.g. binding affinity), map the molecules into 2D space using PCA and visualize the data colored by the property.
- Read SMILES
- Save title as property to label with
- Compute fingerprint
- Convert fingerprint bits into an array of size 1024 of zeroes and ones
- Use
sklearn.decomposition.PCAto transform the fingerprints into 2D coordinates - Plot the coordinates and color by specified property
#fill in fps with fingerprint bits, yvals with title value
pca = PCA(n_components=2)
res = pca.fit_transform(fps)
plt.scatter(res[:,0],res[:,1],c=yvals)
plt.gca().set_aspect('equal', adjustable='box');
!wget http://mscbio2025.csb.pitt.edu/files/er.smi
--2023-10-30 20:19:10-- http://mscbio2025.csb.pitt.edu/files/er.smi Resolving mscbio2025.csb.pitt.edu (mscbio2025.csb.pitt.edu)... 136.142.4.139 Connecting to mscbio2025.csb.pitt.edu (mscbio2025.csb.pitt.edu)|136.142.4.139|:80... connected. HTTP request sent, awaiting response... 200 OK Length: 20022 (20K) [application/smil+xml] Saving to: ‘er.smi’ er.smi 100%[===================>] 19.55K --.-KB/s in 0s 2023-10-30 20:19:10 (38.3 MB/s) - ‘er.smi’ saved [20022/20022]