%%html
<script src="https://bits.csb.pitt.edu/preamble.js"></script>
FASTA¶
First line is description of sequence and starts with >
All lines up to the next > are part of the same sequence
Usually less than 80 characters per line
>gi|568815581:c4949086-4945650 Homo sapiens chromosome 17, GRCh38.p2 Primary Assembly
CCCGCAGGGTCCACACGGGTCGGGCCGGGCGCGCTCCCGTGCAGCCGGCTCCGGCCCCGACCGCCCCATG
CACTCCCGGCCCCGGCGCAGGCGCAGGCGCGGGCACACGCGCCGCCGCCCGCCGGTCCTTCCCTTCGGCG
GAGGTGGGGGAAGGAGGAGTCATCCCGTTTAACCCTGGGCTCCCCGAACTCTCCTTAATTTGCTAAATTT
GCAGCTTGCTAATTCCTCCTGCTTTCTCCTTCCTTCCTTCTTCTGGCTCACTCCCTGCCCCGATACCAAA
GTCTGGTTTATATTCAGTGCAAATTGGAGCAAACCCTACCCTTCACCTCTCTCCCGCCACCCCCCATCCT
TCTGCATTGCTTTCCATCGAACTCTGCAAATTTTGCAATAGGGGGAGGGATTTTTAAAATTGCATTTGCA
Genbank¶
Annotated format. Starts with LOCUS field. Can have several other annotation (e.g. KEYWORDS, SOURCE, REFERENCE, FEATURES).
ORIGIN record indicates start of sequence
'\\' indicates the end of sequence
LOCUS CAG28598 140 aa linear PRI 16-OCT-2008
DEFINITION PFN1, partial [Homo sapiens].
ACCESSION CAG28598
VERSION CAG28598.1 GI:47115277
DBSOURCE embl accession CR407670.1
KEYWORDS .
SOURCE Homo sapiens (human)
ORGANISM Homo sapiens
Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi;
Mammalia; Eutheria; Euarchontoglires; Primates; Haplorrhini;
Catarrhini; Hominidae; Homo.
ORIGIN
1 magwnayidn lmadgtcqda aivgykdsps vwaavpgktf vnitpaevgv lvgkdrssfy
61 vngltlggqk csvirdsllq dgefsmdlrt kstggaptfn vtvtktdktl vllmgkegvh
121 gglinkkcye mashlrrsqy
//
Biopython¶
Biopython features include parsers for various Bioinformatics file formats (BLAST, Clustalw, FASTA, Genbank,...), access to online services (NCBI, Expasy,...), interfaces to common and not-so-common programs (Clustalw, DSSP, MSMS...), a standard sequence class, various clustering modules, a KD tree data structure etc. and even documentation.
Other modules that might be of interest:
- Pycogent: http://pycogent.org/
- bx-python: http://bitbucket.org/james_taylor/bx-python/wiki/Home
- DendroPy: http://packages.python.org/DendroPy/
- Pygr: http://code.google.com/p/pygr/
- bioservices: https://bioservices.readthedocs.io/en/master/
Biopython is not for performing sequencing itself (see: https://crc.pitt.edu/training/fall-2021-next-generation-sequencing-workshops).
Sequence Objects¶
from Bio.Seq import Seq # the submodule structure of biopython is a little awkward
s = Seq("GATTACA")
s
Seq('GATTACA')
Sequences act a lot like strings, but have additional methods.
Methods shared with str: count, endswith, find, lower, lstrip, rfind, split, startswith, strip,upper
Seq methods:back_transcribe, complement, reverse_complement, tomutable, tostring, transcribe, translate, ungap
Accessing Seq data¶
Sequences act like strings (indexed from 0)
s[0]
'G'
s[2:4] #returns sequence
Seq('TT')
s.lower()
Seq('gattaca')
s + s
Seq('GATTACAGATTACA')
The Central Dogma¶
DNA coding strand (aka Crick strand, strand +1)
5’ ATGGCCATTGTAATGGGCCGCTGAAAGGGTGCCCGATAG 3’
|||||||||||||||||||||||||||||||||||||||
3’ TACCGGTAACATTACCCGGCGACTTTCCCACGGGCTATC 5’
DNA template strand (aka Watson strand, strand −1)
|
Transcription
↓
5’ AUGGCCAUUGUAAUGGGCCGCUGAAAGGGUGCCCGAUAG 3’
Single stranded messenger RNA
|
Translation
↓
MAIVMGR*KGAR*
amino acid sequence (w/stop codons)
dna = Seq('GATTACAGATTACAGATTACA')
dna.complement(),dna.reverse_complement()
(Seq('CTAATGTCTAATGTCTAATGT'), Seq('TGTAATCTGTAATCTGTAATC'))
The Central Dogma¶
dna
Seq('GATTACAGATTACAGATTACA')
rna = dna.transcribe()
rna
Seq('GAUUACAGAUUACAGAUUACA', RNAAlphabet())
protein = rna.translate()
protein
Seq('DYRLQIT', ExtendedIUPACProtein())
dna.translate() #unlike cells, don't actually need rna
Seq('DYRLQIT', ExtendedIUPACProtein())
Codon Tables¶
By default the standard translation table is used, but others can be provided to the translate method.
from Bio.Data import CodonTable
print(sorted(CodonTable.unambiguous_dna_by_name.keys()))
['Alternative Flatworm Mitochondrial', 'Alternative Yeast Nuclear', 'Archaeal', 'Ascidian Mitochondrial', 'Bacterial', 'Balanophoraceae Plastid', 'Blastocrithidia Nuclear', 'Blepharisma Macronuclear', 'Candidate Division SR1', 'Chlorophycean Mitochondrial', 'Ciliate Nuclear', 'Coelenterate Mitochondrial', 'Condylostoma Nuclear', 'Dasycladacean Nuclear', 'Echinoderm Mitochondrial', 'Euplotid Nuclear', 'Flatworm Mitochondrial', 'Gracilibacteria', 'Hexamita Nuclear', 'Invertebrate Mitochondrial', 'Karyorelict Nuclear', 'Mesodinium Nuclear', 'Mold Mitochondrial', 'Mycoplasma', 'Pachysolen tannophilus Nuclear', 'Peritrich Nuclear', 'Plant Plastid', 'Protozoan Mitochondrial', 'Pterobranchia Mitochondrial', 'SGC0', 'SGC1', 'SGC2', 'SGC3', 'SGC4', 'SGC5', 'SGC8', 'SGC9', 'Scenedesmus obliquus Mitochondrial', 'Spiroplasma', 'Standard', 'Thraustochytrium Mitochondrial', 'Trematode Mitochondrial', 'Vertebrate Mitochondrial', 'Yeast Mitochondrial']
print(CodonTable.unambiguous_dna_by_name['Standard'])
Table 1 Standard, SGC0 | T | C | A | G | --+---------+---------+---------+---------+-- T | TTT F | TCT S | TAT Y | TGT C | T T | TTC F | TCC S | TAC Y | TGC C | C T | TTA L | TCA S | TAA Stop| TGA Stop| A T | TTG L(s)| TCG S | TAG Stop| TGG W | G --+---------+---------+---------+---------+-- C | CTT L | CCT P | CAT H | CGT R | T C | CTC L | CCC P | CAC H | CGC R | C C | CTA L | CCA P | CAA Q | CGA R | A C | CTG L(s)| CCG P | CAG Q | CGG R | G --+---------+---------+---------+---------+-- A | ATT I | ACT T | AAT N | AGT S | T A | ATC I | ACC T | AAC N | AGC S | C A | ATA I | ACA T | AAA K | AGA R | A A | ATG M(s)| ACG T | AAG K | AGG R | G --+---------+---------+---------+---------+-- G | GTT V | GCT A | GAT D | GGT G | T G | GTC V | GCC A | GAC D | GGC G | C G | GTA V | GCA A | GAA E | GGA G | A G | GTG V | GCG A | GAG E | GGG G | G --+---------+---------+---------+---------+--

%%html
<div id="seqtrans" style="width: 500px"></div>
<script>
$('head').append('<link rel="stylesheet" href="https://bits.csb.pitt.edu/asker.js/themes/asker.default.css" />');
var divid = '#seqtrans';
jQuery(divid).asker({
id: divid,
question: "What amino acid does the codon <tt>CAT</tt> translate to?",
answers: ['H',"Y","V","A","X"],
extra: ['Histidine','Tyrosine','Valine','Alanine','Other'],
server: "https://bits.csb.pitt.edu/asker.js/example/asker.cgi",
charter: chartmaker})
$(".jp-InputArea .o:contains(html)").closest('.jp-InputArea').hide();
</script>
SeqRecord¶
Sequence data is read/written as SeqRecord objects.
These objects store additional information about the sequence (name, id, description, features)
SeqIO reads sequence records:
- must specify format
readto read a file with a single recordparseto iterate over file with mulitple records
from Bio.SeqRecord import SeqRecord
from Bio import SeqIO
seq = SeqIO.read('../files/p53.gb','genbank')
seq
SeqRecord(seq=Seq('GATGGGATTGGGGTTTTCCCCTCCCATGTGCTCAAGACTGGCGCTAAAAGTTTT...GTG', IUPACAmbiguousDNA()), id='NG_017013.2', name='NG_017013', description='Homo sapiens tumor protein p53 (TP53), RefSeqGene (LRG_321) on chromosome 17', dbxrefs=[])
seqs = []
# https://MSCBIO2025.github.io/files/hydra.fasta
for s in SeqIO.parse('../files/hydra.fasta','fasta'):
seqs.append(s)
len(seqs)
40
Fetching sequences from the Internet¶
Biopython provides and interface to the NCBI "Entrez" search engine
The results of internet queries are returned as file-like objects.
from Bio import Entrez
Entrez.email = "jpbarton@pitt.edu" # biopython forces you to provide your email
res = Entrez.read(Entrez.einfo()) # the names of all available databases
res
{'DbList': ['pubmed', 'protein', 'nuccore', 'ipg', 'nucleotide', 'structure', 'genome', 'annotinfo', 'assembly', 'bioproject', 'biosample', 'blastdbinfo', 'books', 'cdd', 'clinvar', 'gap', 'gapplus', 'grasp', 'dbvar', 'gene', 'gds', 'geoprofiles', 'homologene', 'medgen', 'mesh', 'nlmcatalog', 'omim', 'orgtrack', 'pmc', 'popset', 'proteinclusters', 'pcassay', 'protfam', 'pccompound', 'pcsubstance', 'seqannot', 'snp', 'sra', 'taxonomy', 'biocollections', 'gtr']}
print(sorted(res['DbList']))
['annotinfo', 'assembly', 'biocollections', 'bioproject', 'biosample', 'blastdbinfo', 'books', 'cdd', 'clinvar', 'dbvar', 'gap', 'gapplus', 'gds', 'gene', 'genome', 'geoprofiles', 'grasp', 'gtr', 'homologene', 'ipg', 'medgen', 'mesh', 'nlmcatalog', 'nuccore', 'nucleotide', 'omim', 'orgtrack', 'pcassay', 'pccompound', 'pcsubstance', 'pmc', 'popset', 'protein', 'proteinclusters', 'protfam', 'pubmed', 'seqannot', 'snp', 'sra', 'structure', 'taxonomy']
ESearch¶
You can search any database for a given term and it will return the ids of all the relevant records.
result = Entrez.esearch(db='nucleotide', term='tp53') # the result is a file-like object of the raw xml data
records = Entrez.read(result) # put into a more palatable form (dictionary)
print(records)
{'Count': '16953', 'RetMax': '20', 'RetStart': '0', 'IdList': ['2579134893', '2577097206', '1675046266', '1621312268', '1621310586', '1621309831', '1565520846', '1565520832', '1519314995', '1519314531', '1519245996', '1519245289', '1492459888', '2544609391', '2544608770', '2544608358', '2544608205', '1732746165', '2578505094', '2578491630'], 'TranslationSet': [], 'TranslationStack': [{'Term': 'tp53[All Fields]', 'Field': 'All Fields', 'Count': '16953', 'Explode': 'N'}, 'GROUP'], 'QueryTranslation': 'tp53[All Fields]'}
There were 2923 7616 8238 10725 11850 12751 13952 16943 hits, but by default only 20 are returned. We can change this (and other parameters) by changing our search terms.
records = Entrez.read(Entrez.esearch(db='nucleotide', term='tp53', retmax=50))
records
{'Count': '16953', 'RetMax': '50', 'RetStart': '0', 'IdList': ['2579134893', '2577097206', '1675046266', '1621312268', '1621310586', '1621309831', '1565520846', '1565520832', '1519314995', '1519314531', '1519245996', '1519245289', '1492459888', '2544609391', '2544608770', '2544608358', '2544608205', '1732746165', '2578505094', '2578491630', '2544608591', '1677501386', '1677500090', '1653961881', '349585153', '50344732', '2577326767', '2577319026', '2577314085', '2577314083', '2577314081', '2577144630', '2577144628', '2577144626', '2577144624', '2577144622', '2577144620', '2577144618', '2577112865', '2577109742', '2577244898', '2577244896', '2577244894', '2577238979', '2577226876', '2577220539', '2577220537', '2577179306', '2577179304', '2577176327'], 'TranslationSet': [], 'TranslationStack': [{'Term': 'tp53[All Fields]', 'Field': 'All Fields', 'Count': '16953', 'Explode': 'N'}, 'GROUP'], 'QueryTranslation': 'tp53[All Fields]'}
EFetch¶
To get the full record for a given id, use efetch.
Must provide rettype (available options include fasta and gb)
retmode can be text or xml.
#fetch the genbank file for the first id from our search
result = Entrez.efetch(db="nucleotide",id=records['IdList'][0],rettype="gb",retmode='text')
#parse into a seqrecord
p53 = SeqIO.read(result,'gb')
result
<_io.TextIOWrapper encoding='latin-1'>
p53
SeqRecord(seq=Seq('TCAGGGTCCAGGTGATTCTGAACGAGCTGAAAATCGAAGATAAACAACACTTTC...AAA', IUPACAmbiguousDNA()), id='XM_059624316.1', name='XM_059624316', description='PREDICTED: Neocloeon triangulifer TP53-regulated inhibitor of apoptosis 1-like (LOC132199519), mRNA', dbxrefs=['BioProject:PRJNA1017488'])
Features¶
Genbank files are typically annotated with features, which refer to portions of the full sequence
SeqRecord objects track these features and you can extract the corresponding subsequence
CDS - coding sequence
p53.features
[SeqFeature(FeatureLocation(ExactPosition(0), ExactPosition(712), strand=1), type='source'), SeqFeature(FeatureLocation(ExactPosition(0), ExactPosition(712), strand=1), type='gene'), SeqFeature(FeatureLocation(ExactPosition(197), ExactPosition(437), strand=1), type='CDS'), SeqFeature(FeatureLocation(ExactPosition(711), ExactPosition(712), strand=1), type='polyA_site')]
Extracting subsequences¶
cdsfeature = p53.features[2]
print(cdsfeature)
type: CDS
location: [197:437](+)
qualifiers:
Key: codon_start, Value: ['1']
Key: db_xref, Value: ['GeneID:132199519']
Key: gene, Value: ['LOC132199519']
Key: product, Value: ['TP53-regulated inhibitor of apoptosis 1-like']
Key: protein_id, Value: ['XP_059480299.1']
Key: translation, Value: ['MNSVGEECNDMKKTYDSCFNAWFSHKFLRGQHDDQMCADLFLKYQQCVKNAMKQQNISIEDVQHFQLGTEEEKKAPPKK']
The subsequence the feature refers to can be extracted from the original full sequence using the feature.
coding = cdsfeature.extract(p53) #pass the full record (p53) to the feature
coding
SeqRecord(seq=Seq('ATGAACAGTGTCGGCGAGGAATGCAACGACATGAAAAAAACTTATGATTCCTGC...TAG', IUPACAmbiguousDNA()), id='XM_059624316.1', name='XM_059624316', description='PREDICTED: Neocloeon triangulifer TP53-regulated inhibitor of apoptosis 1-like (LOC132199519), mRNA', dbxrefs=[])
BLAST!¶
Biopython let's you use NCBI's BLAST to search for similar sequences with qblast which has three required arguments:
- program: blastn, blastp, blastx, tblastn, tblastx
- database: see website
- sequence: a sequence object
from Bio.Blast import NCBIWWW
result = NCBIWWW.qblast("blastn","nt",coding.seq,hitlist_size=250)
#result is a file-like object with xml in it - it can take a while to get results
from Bio.Blast import NCBIXML #for parsing xmls
blast_records = NCBIXML.read(result)
print(len(blast_records.alignments),len(blast_records.descriptions))
147 147
alignment = blast_records.alignments[0]
print(len(alignment.hsps))
1
hsp = alignment.hsps[0] # high scoring segment pairs
print('****Alignment****')
print('sequence:', alignment.title)
print('length:', alignment.length)
print('e value:', hsp.expect)
print(hsp.query[0:75] + '...') # what we searched with
print(hsp.match[0:75] + '...')
print(hsp.sbjct[0:75] + '...') # what we matched to
****Alignment**** sequence: gi|2489302823|emb|OX463782.1| Siphlonurus alternatus genome assembly, chromosome: 5 length: 40728708 e value: 2.75518e-13 ATGAACAGTGTCGGCGAGGAATGCAACGACATGAAAAAAACTTATGATTCCTGCTTCAACGCATGGTTCAGCCAC... ||||| ||||| || || || || | || ||||| || ||||||||||| || | ||||| ||||| ... ATGAATAGTGTTGGAGAAGAGTGTACTGATATGAAGAAGCAATATGATTCCTGTTTTCATCTTTGGTTTAGCCAA...
alignment = blast_records.alignments[-1] # get last alignment
hsp = alignment.hsps[0]
print('****Alignment****')
print('sequence:', alignment.title)
print('length:', alignment.length)
print('e value:', hsp.expect)
print(hsp.query[0:75] + '...') # what we searched with
print(hsp.match[0:75] + '...')
print(hsp.sbjct[0:75] + '...') # what we matched to
****Alignment**** sequence: gi|2362810685|emb|OX371006.1| Yponomeuta malinellus genome assembly, chromosome: 12 length: 19760461 e value: 2.94431 AGAATGCAATGAAGCAGCAAAACAT... |||||||||||||||||||||||||... AGAATGCAATGAAGCAGCAAAACAT...
Creating alignments¶
Biopython uses clustal to perform multiple alignments
The interface is very simple - you construct the clustal commandline and must read the result files
from Bio.Align.Applications import ClustalwCommandline
cline = ClustalwCommandline('clustalw', infile='../files/hydra.fasta',outfile='alignment.aln')
print(cline)
clustalw -infile=../files/hydra.fasta -outfile=alignment.aln
output = cline() #this calls the above, may take a while # If it doesn't work, sudo apt install clustalw
!head alignment.aln
CLUSTAL 2.1 multiple sequence alignment gi|164609123|gb|ABY62783.1|/1- ------------MLNIAILCLSCYINYVSCLSLTSPAIIEEVVGRSVTIT model1_F.pdb ------------------------------LSLTSPAIIEEVVGRSVTIT gi|225423262|gb|ACN91137.1|/1- ------------MLNIAILSLSCYINYVSCLSLTSPAIIEEVVGRSVTIT gi|302171750|gb|ADK97776.1|/1- ------------MLNIAILSLSCYINYVSCLSLTSPAIIEEVVGRSVTIT gi|302171774|gb|ADK97788.1|/1- ------------MLNIAILCLSCYINYVSCLSLTSSAIIEEVVNRSVTIT gi|225423244|gb|ACN91128.1|/1- ------------MLNIAILCLSCYINYVSCLSLTSSAIIEEVVNRSVTIT gi|225423272|gb|ACN91142.1|/1- ------------MLNIAILLLSCYINYVSSLSLTSPAIIEEVVNRSVTIT
Alignments¶
AlignIO is used to read alignment files (must provide format)
from Bio import AlignIO
align = AlignIO.read('alignment.aln','clustal')
align
<<class 'Bio.Align.MultipleSeqAlignment'> instance (40 records of length 691, SingleLetterAlphabet()) at 1068300a0>
print(align)
SingleLetterAlphabet() alignment with 40 rows and 691 columns ------------MLNIAILCLSCYINYVSCLSLTSPAIIEEVVG...QKK gi|164609123|gb|ABY62783.1|/1- ------------------------------LSLTSPAIIEEVVG...--- model1_F.pdb ------------MLNIAILSLSCYINYVSCLSLTSPAIIEEVVG...QKK gi|225423262|gb|ACN91137.1|/1- ------------MLNIAILSLSCYINYVSCLSLTSPAIIEEVVG...QKK gi|302171750|gb|ADK97776.1|/1- ------------MLNIAILCLSCYINYVSCLSLTSSAIIEEVVN...QKK gi|302171774|gb|ADK97788.1|/1- ------------MLNIAILCLSCYINYVSCLSLTSSAIIEEVVN...QKK gi|225423244|gb|ACN91128.1|/1- ------------MLNIAILLLSCYINYVSSLSLTSPAIIEEVVN...QKK gi|225423272|gb|ACN91142.1|/1- ------------MLNIAILCLSYYVNYVSCLSLTSPAIIEEVVG...QKK gi|302171742|gb|ADK97772.1|/1- ------------MLNIAILCLSYYVNYVSCLSLTSPAIIEEVVG...QKK gi|225423264|gb|ACN91138.1|/1- ------------MLNIAILSLSCYINYVSCLSLTSPAIIEEVVG...QKK gi|302171780|gb|ADK97791.1|/1- ------------MLNIAILCLSCYINYVSCVSLTSPAIIEEVVG...QKK gi|302171756|gb|ADK97779.1|/1- ------------MLNVVILCLSCYINYVSCLSLTSPAIIEEVVG...QKK gi|225423288|gb|ACN91150.1|/1- MKLVIVIPNDENLLNIAILCLSCYLNYVSCLSLTSPAIIEKVVG...QKK gi|225423270|gb|ACN91141.1|/1- ------------MLNIAILCLSCYINYVSCVSVTSPPIIEEVVG...QKK gi|302171766|gb|ADK97784.1|/1- ------------MLNIAILCLSCYINYVSCVSVTSPPIIEEVVG...QKK gi|225423256|gb|ACN91134.1|/1- ------------MLNIAILCLSCYINYVSCVSVTSPPIIEEVVG...QKK gi|302171748|gb|ADK97775.1|/1- ------------MLNIAILCLSCYINYVSCLSLTSSAIIEEVVG...QKK gi|302171772|gb|ADK97787.1|/1- ------------MLNIAILCLSCYINYVSCLSVTSPAIIEEVVG...QKK gi|225423294|gb|ACN91153.1|/1- ... ------------MLNIAILSLSCYINYVSCLSLTSPAIIEKVVG...QKK gi|302171764|gb|ADK97783.1|/1-
Slicing Alignments¶
Alignments are sliced just like numpy arrays
align[0] #first row
SeqRecord(seq=Seq('------------MLNIAILCLSCYINYVSCLSLTSPAIIEEVVGRSVTITYVTD...QKK', SingleLetterAlphabet()), id='gi|164609123|gb|ABY62783.1|/1-', name='<unknown name>', description='gi|164609123|gb|ABY62783.1|/1-', dbxrefs=[])
align[:,0] #first column
'------------M-----------------M---------'
print(align[:,0:40])
SingleLetterAlphabet() alignment with 40 rows and 40 columns ------------MLNIAILCLSCYINYVSCLSLTSPAIIE gi|164609123|gb|ABY62783.1|/1- ------------------------------LSLTSPAIIE model1_F.pdb ------------MLNIAILSLSCYINYVSCLSLTSPAIIE gi|225423262|gb|ACN91137.1|/1- ------------MLNIAILSLSCYINYVSCLSLTSPAIIE gi|302171750|gb|ADK97776.1|/1- ------------MLNIAILCLSCYINYVSCLSLTSSAIIE gi|302171774|gb|ADK97788.1|/1- ------------MLNIAILCLSCYINYVSCLSLTSSAIIE gi|225423244|gb|ACN91128.1|/1- ------------MLNIAILLLSCYINYVSSLSLTSPAIIE gi|225423272|gb|ACN91142.1|/1- ------------MLNIAILCLSYYVNYVSCLSLTSPAIIE gi|302171742|gb|ADK97772.1|/1- ------------MLNIAILCLSYYVNYVSCLSLTSPAIIE gi|225423264|gb|ACN91138.1|/1- ------------MLNIAILSLSCYINYVSCLSLTSPAIIE gi|302171780|gb|ADK97791.1|/1- ------------MLNIAILCLSCYINYVSCVSLTSPAIIE gi|302171756|gb|ADK97779.1|/1- ------------MLNVVILCLSCYINYVSCLSLTSPAIIE gi|225423288|gb|ACN91150.1|/1- MKLVIVIPNDENLLNIAILCLSCYLNYVSCLSLTSPAIIE gi|225423270|gb|ACN91141.1|/1- ------------MLNIAILCLSCYINYVSCVSVTSPPIIE gi|302171766|gb|ADK97784.1|/1- ------------MLNIAILCLSCYINYVSCVSVTSPPIIE gi|225423256|gb|ACN91134.1|/1- ------------MLNIAILCLSCYINYVSCVSVTSPPIIE gi|302171748|gb|ADK97775.1|/1- ------------MLNIAILCLSCYINYVSCLSLTSSAIIE gi|302171772|gb|ADK97787.1|/1- ------------MLNIAILCLSCYINYVSCLSVTSPAIIE gi|225423294|gb|ACN91153.1|/1- ... ------------MLNIAILSLSCYINYVSCLSLTSPAIIE gi|302171764|gb|ADK97783.1|/1-
And now for a brief foray into marine microbiology...¶

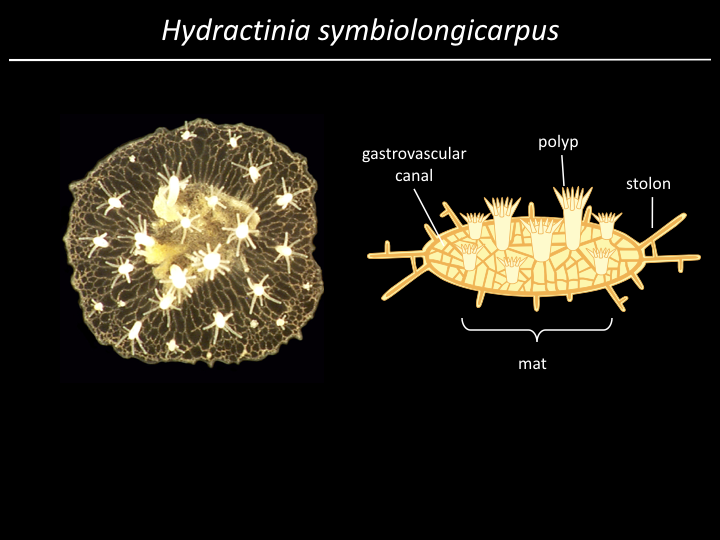
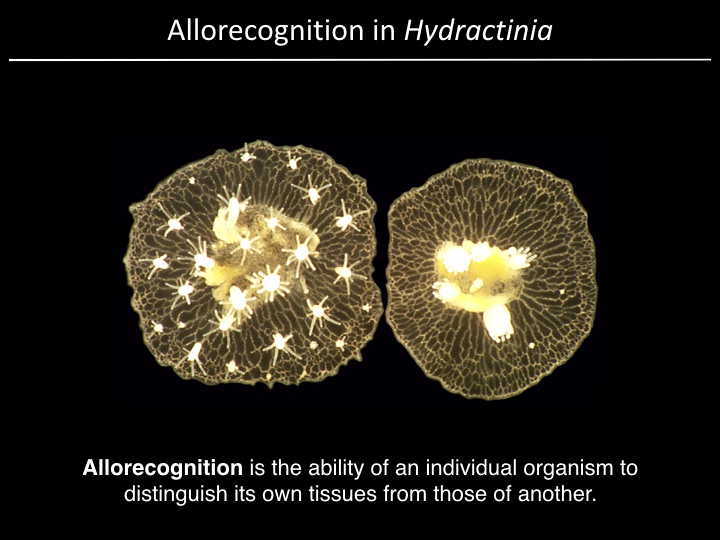

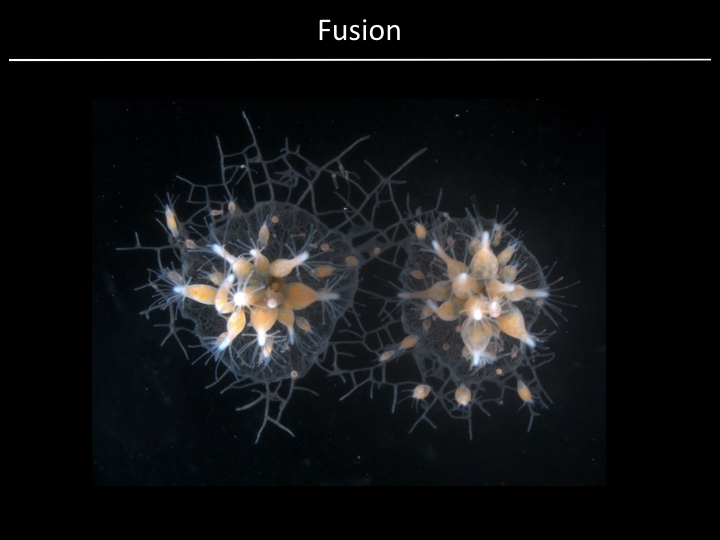
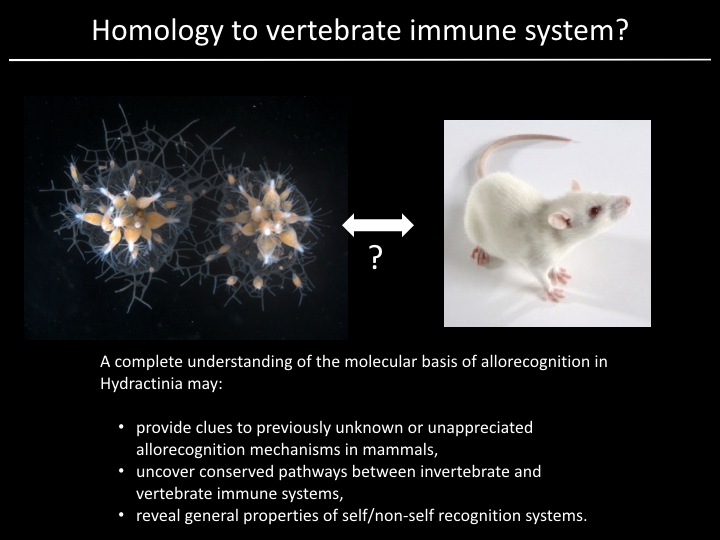
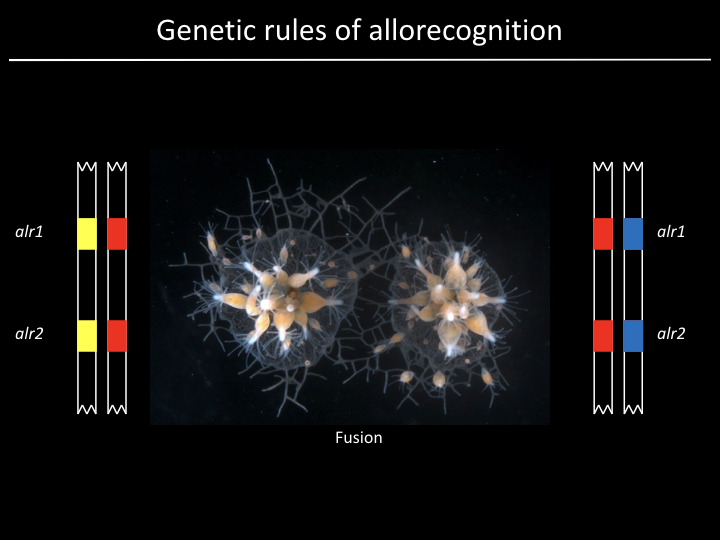
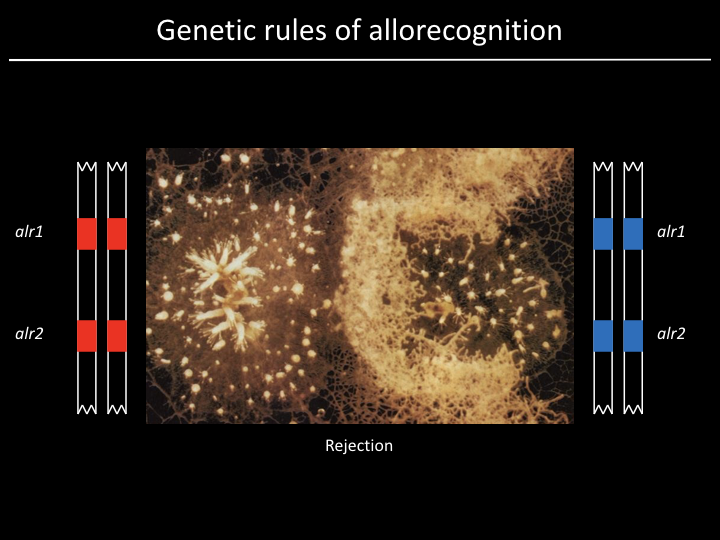
Project¶
We have a gene (alr2), but what part of the gene is responsible for allorecognition?
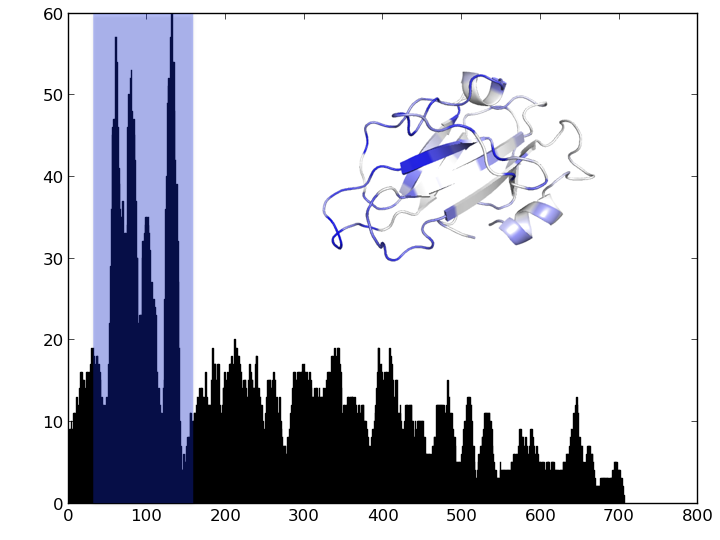
Given 179 sequences, how might we find out?
Find the part of the sequence that changes the most.
- Count number of distinct residues at each position. Plot.
- Count number of distinct subsequences of length 10 at each position. Plot.
!wget https://MSCBIO2025.github.io/files/hydra179.aln
--2023-09-21 12:27:54-- https://mscbio2025.github.io/files/hydra179.aln Resolving mscbio2025.github.io (mscbio2025.github.io)... 185.199.110.153, 185.199.109.153, 185.199.108.153, ... Connecting to mscbio2025.github.io (mscbio2025.github.io)|185.199.110.153|:443... connected. HTTP request sent, awaiting response... 301 Moved Permanently Location: https://mscbio2025.net/files/hydra179.aln [following] --2023-09-21 12:27:54-- https://mscbio2025.net/files/hydra179.aln Resolving mscbio2025.net (mscbio2025.net)... 185.199.111.153, 185.199.108.153, 185.199.109.153, ... Connecting to mscbio2025.net (mscbio2025.net)|185.199.111.153|:443... connected. HTTP request sent, awaiting response... 200 OK Length: 218936 (214K) [application/octet-stream] Saving to: ‘hydra179.aln’ hydra179.aln 100%[===================>] 213.80K --.-KB/s in 0.01s 2023-09-21 12:27:55 (16.1 MB/s) - ‘hydra179.aln’ saved [218936/218936]